

Research Article
Bioequivalence, Safety and Tolerability of Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged-Release Tablets in Healthy Adult Volunteers
Vamshi Ramana Prathap, Harikiran Chary Vadla, Balaramesha Chary Rallabandi and Ramesh Gannu*
AET Laboratories Pvt. Ltd., Survey No. 42, Gaddapotharam Village Kazipally Industrial Area, Hyderabad, Telangana State, India
*Address for Correspondence: Ramesh Gannu, Assist General Manager, Product Development, AET Laboratories Pvt. Ltd., Survey No. 42, Gaddapotharam Village Kazipally Industrial Area, Hyderabad, Telangana State, India, ORCID ID: orcid.org/0000-0002-3259-3053; Tel +914-039-102-936/38; Fax +914-039-102-931; E-mail: g.ramesh@aet.in
Dates: 07 September 2017; Approved: 21 September 2017; Published: 22 September 2017
Citation this article: Vamshi RP, Harikiran CV, Balaramesha CR, Ramesh G. Bioequivalence, Safety and Tolerability of Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged-Release Tablets in Healthy Adult Volunteers. Am J Pharmacol Ther. 2018;2(1): 001-106.
Copyright: © Ramesh G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Sitagliptin; Metformin; Pharmacokinetics; Bioequivalence; Safety; Tolerability
Abstract
The objective of the study was to evaluate the bioequivalence, safety and tolerability of a fixed-dose combination formulation, FDC (Sitagliptin 100 mg and Metformin hydrochloride 1000 mg prolonged-release tablets) relative to reference formulations (Januvia® 100 mg Filmtabletten co-administered with Glucophage® SR 1000 mg Prolonged-release Tablets) after single dose administration under fasted and fed condition in healthy adult male and female volunteers. An open label, two period, two treatment, two-way crossover, randomized study design was utilized for the investigation. Blood samples were collected at scheduled time points till 120 h after drug administration. A wash-out period of 14 days was maintained between two periods. Eight adverse events of mild in severity occurred in five subjects in the study; no serious adverse events reported.
The statistical analysis of the pharmacokinetics indicates that FDC met the bioequivalence criteria under fed state for both drugs. While Sitagliptin was bioequivalent in fasted state and Metformin showed indicative results, possibly could show bioequivalence in adequate number of subjects.
The conclusions drawn from the study indicate that, FDC was tolerable and the adverse events are similar to the reference products and are mild in nature, hence safe to use in human subjects. FDC showed comparable in vivo behavior to that of reference formulations.
Introduction
Sitagliptin, a dipeptidyl peptidase 4 (DPP-4) inhibitor indicated for hyperglycemia [1]. Sitagliptin exerts its action by prolonging the action of GLP-1 (glucagon-like peptide-1) and GIP (glucose-dependent insulin tropic polypeptide) also increases insulin production and lowers glucagon secretion from pancreatic alpha cells, which decreases hepatic glucose overproduction [2]. The enzyme, DPP-4 rapidly hydrolyzes the incretin hormones [3]. Sitagliptin inhibits the enzyme, DPP-4 and thereby the action of incretin hormones is prolonged. Sitagliptin shows a quicker absorption post oral administration with a Tmax of 1-4 h [4]. Food does not show a significant influence on the pharmacokinetics of Sitagliptin, hence Sitagliptin can be taken without regards to food.
Metformin, is prescribed as first line therapy in type-2 diabetes [5] Metformin inhibits the processes (gluconeogenesis and glycogenolysis) by which glucose is formed in liver; improving insulin action; utilization of glucose and by delay of intestinal glucose absorption. Prolonged release formulation of Metformin, shows peak plasma concentration at about 7 h after oral administration [6].
A FDC comprising of Sitagliptin 100 mg and Metformin hydrochloride 1000 mg prolonged release was developed. FDCs offers numerous merits in comparison to individual drug products that include [7], the dosage forms are simple in terms of dosage schedule which improves patient compliance and therefore improves the outcome of treatment. This is especially important in elderly patients or patients suffering from multiple disorders. The FDCs can reduce ‘treatment burden’, therefore can greatly increase the overall outcome of treatment.
The aim of the present paper is to describe the outcome of study designed to assess comparative pharmacokinetics, safety and tolerability of a newly developed oral test formulation (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged-release tablets) against individual reference products. The reference products are registered and marketed products, include Januvia® (Sitagliptin) 100 mg Filmtabletten and Glucophage® (Metformin) SR 1000 mg Prolonged-release Tablets. The products were evaluated in healthy, adult volunteers under fasting and fed conditions in accordance to the EMA and other applicable regulatory requirements [8].
Methods
Study subjects: inclusion and exclusion criteria
Twenty four healthy male and female volunteers were enrolled in the study (Table 1), each cohort comprises of 12 subjects (Table 2). Subjects underwent for a screening procedure within 7 days prior to entering the study. All subjects were provided with a volunteer information leaflet and written informed consent to participate in the study. The signed copies of consents were obtained from all the subjects prior to enrolment. The subjects were free to withdraw their participation at any time during the entire study. Subjects were eligible to participate if aged 18 years (subject must have turned 18 prior to screening) or older with a Body Mass Index (BMI) ≥18.5 and ≤30 kg/m2 [9]. Furthermore subjects needed to be healthy volunteers as per screening criteria with a willingness to sign the informed consent form and to adhere to protocol throughout the study. Non-smokers or ex-smokers were enrolled in the study. Subjects needed to agree to abstain from alcohol 48 hours before initiation of study and during each study period. Female healthy volunteers who agree to use of an effective non-hormonal method of contraception or abstinence starting from screening and until follow-up examination are only enrolled.
Subjects were excluded from the study based on history of hypersensitivity to the test drug or to drugs belonging to the same pharmacological and chemical class. Subjects with history or presence of any relevant medical condition including cancer, significant disease of the renal, hepatic, gastrointestinal, respiratory, cardiovascular, endocrine or locomotor systems and any metabolic, hematological or neurological disorder were excluded from the study. Subject with heart rate outside the normal range of 50-100 beats per minute or a body temperature outside the normal range of 35.5-37.4°C or a respiratory rate outside the normal range of 14-20 breaths per minute or a sitting blood pressure less than 90/ 60 mmHg or more than 140/ 90 mmHg at the screening examination were not included in the study. Furthermore ECG evidence of any clinically significant abnormalities, any recent history (within the last two years) of drug or alcohol abuse, recent psychiatric disorder or use of psychotropic medicines, or positive results to the HIV or hepatitis C or hepatitis B tests led to non-enrolment of the subjects. Breast feeding females or female subjects with a positive pregnancy test were excluded from enrolment.
Ethical consideration
The study protocol was approved by National Ethics Committee for Drugs Clinical Trials and to the Medicines and Medical Devices Agency, Chisinau, The Moldavian Republic. The study was conducted in agreement with the Declaration of Helsinki (1964 and following amendments), ICH-GCP R2 (2017), EEC rules concerning human experimentation (No. 91/507/EEC) and Directive 2001/20/EC of The European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulation and administrative provisions of the Member States relating to the implement of good clinical practice in the conduct of clinical trials on medicinal products for human use.
Medical examination
Before enrolment, each volunteer was interviewed by the Clinical Investigator for medical history. A medical examination was conducted to record SAP (Systolic Arterial Pressure), DAP (Diastolic Arterial Pressure), Heart Rate (HR), ECG, body temperature and respiratory frequency. Urine and venous blood samples have been obtained for urinalysis, routine hematology, serology and clinical chemistry testing respectively.
Formulations tested
The fixed dose combination (FDC, composition is not disclosed) containing Sitagliptin 100 mg and Metformin hydrochloride 1000 mg prolonged-release tablets (AET Laboratories Pvt. Ltd, India) was used as test formulation. The in vivo behavior of test formulation for two drugs was compared with individual reference formulations namely Januvia® 100 mg filmtabletten and Glucophage® SR 1000 mg prolonged-release tablets (both manufactured by Merck Sharp & Dohme Ltd., UK). The details of formulations administered with respect to Cohort was shown in table 2. The formulations were dosed in two Cohorts. In Cohort 1, the formulations were administered on an empty stomach (after 10 hours of overnight fasting). In Cohort 2, the formulations were administered under fed conditions. A standard high-fat, high-calories breakfast (composed of 25 grams of bread, 10 grams of butter, 2 hard-boiled eggs, 25 grams of bacon, 125 g French fries and 250 ml of whole milk) was served to the volunteers at precisely 30 minutes prior to the scheduled administration time in Cohort 2.
Study design
The study was designed as open label, two-period, two-way cross-over, controlled randomized, single dose comparative bioequivalence study between FDC and reference formulations in healthy, adult male and female volunteers. A wash-out period of fourteen days was followed. The subjects were admitted at the clinical center in the evening prior to each study treatment at about 6 PM and housed until 24.00 hours post dose administration in each period. A standard meals at 6, 9 and 12 hours post-dosing were served on the treatment days for all subjects. No other food was consumed during confinement. Free access to water was granted until 1 hour before dosing and subjects were not allowed to drink water (or any other liquids) until 1 hour after dosing, except the 200 ml water received with study medication and the 250 ml of whole milk served with the standardized high-calorie, high-fat breakfast. Thereafter, water was provided ad libitum, at room temperature. The tablets were swallowed whole, without being broken, chewed or crushed. After administration, a mouth check was performed in order to ensure that the dose has been taken entirely. The volunteers kept a standing position during the treatment intake and stayed in their beds in semi-reclined position for the next 4 hours following dosing.
Blood sample collection, separation and storage
Blood samples were collected at scheduled time points given in the protocol, before dose (5 mL + reserve blank sample 20 mL, only in Period 1) and at 0.33, 0.67, 1.00, 1.33, 1.67, 2.00, 2.33, 2.67, 3.00, 3.33, 3.67, 4.00, 4.33, 4.67, 5.00, 5.33, 5.67, 6.00, 7.00, 8.00, 9.00, 10.00, 12.00, 16.00, 24.00, 48.00, 72.00, 96.00 and 120.00 h. Blood sampling schedule was planned to provide an adequate estimation of Cmax and to cover the concentration-time curve long enough to provide a reliable estimate of extent of absorption. The blood samples were centrifuged for 10 minutes at 4°C nominal with a force of 1500 (±5)-g. After centrifugation, plasma was separated into two aliquots. The frozen samples were stored at -20°C or cooler until shipped to the analytical laboratory.
Method of sample analysis
The identification and quantification of Sitagliptin and Metformin in plasma was performed by Liquid Chromatography/Mass Spectrometry. The analytical method was validated before the start of the analysis of the plasma samples. The analyst was kept blind in respect of the treatment assigned.
Pharmacokinetic variables
The primary pharmacokinetic parameters of Cmax (peak drug concentration) and AUCt (area under the curve from time zero to t) were calculated for Sitagliptin and Metformin. In addition, other secondary parameters such as Tmax (time of the peak drug concentration), AUC0-inf (area under the curve from time zero to infinity) T1/2 (plasma half-life) and MRT (mean residence time) were also calculated.
Statistical analysis
The comparative statistics of the pharmacokinetic data was performed by comparing the fixed dose combination vs respective reference formulation in each Cohort. All statistical calculations were performed using SAS version 9.4 software. Descriptive statistics was done for all pharmacokinetic parameters.
Bioequivalence comparisonThe confidence interval for the ratio of the population means (Test/Reference) was calculated considering a classic 90% confidence interval. The two one-sided parametric t-test according to Schuirmann (ln-transformed values) were applied with the null hypothesis of bioequivalence at the 5% significance level. The bioequivalence acceptance interval was set to 80-125% for Metformin and Sitagliptin.
Safety and tolerability assessment
The clinical safety was assessed via medical history, clinical examination (physical and systemic examination), 12-lead ECG and vital signs (blood pressure, heart rate, and respiratory rate and body temperature) and laboratory parameters (hematology, clinical chemistry and serological parameters) at the time of screening. The parameters were measured soon after reaching the study center, prior to the dosing day of each study period. In the morning before dosing, on each period subjects were asked regarding their well-being and eventual intake of other medication. During the study treatment days (Period 1 and 2), SAP, DAP, HR and body temperature were checked at rest, before drug administration, in laying position. Also during the study days (Day 1 of Period 1 and 2), blood glucose levels were determined before drug administration and at 1.5, 3.0, 4.0, 6.0, 8.0, 12.0, 16.0 and 24.0 hours post dose.
To prevent hypoglycemia, 240 ml of water containing 20% glucose was given to all subjects at 1.0 hour post-dose. Throughout the study period, volunteers were questioned on adverse events occurrence. The second study period started after a washout period of 14 days and the same procedures were followed as for the first study period. Within 2 weeks after the last treatment (actual time: 7 days from the last treatment), all subjects underwent a study exit examination that consisted of the same medical examinations as for the screening, and the same hematological and chemical chemistry and urinalysis laboratory procedures as for the screening. Pregnancy urine test for female subjects were done.
Results
Study subjects
Out of thirty eight adult healthy male and female volunteers screened, a total of twenty four subjects were enrolled in the study and the demographics of the subjects participated in the study was shown in table 1.
| Table 1: Summary statistic on the demographic data. | ||
| Parameter | Mean ± SD (n = 24) | Min-Max |
| Age (years) | 34.71 ± 10.78 | 18-57 |
| Weight (kg) | 66.96 ± 10.91 | 51-94 |
| Height (cm) | 167.75 ± 8.17 | 150-180 |
| BMI (Kg/m2) | 23.74 ± 2.99 | 18.6-29.7 |
Pharmacokinetic and statistical analysis
The mean plasma profiles for Sitagliptin and Metformin from two Cohorts have shown in figure 1 and figure 2. The pharmacokinetic and statistical parameters were analyzed for Sitagliptin and Metformin from two cohorts for the subject who completed both periods of the study. The mean plasma profiles from two cohorts appear to be superimposable for reference and FDC for each component indicating that Sitagliptin and Metformin from FDC and reference formulations have comparable in vivo behavior. There was no significant period or sequence effect found for Cmax, AUCt and AUC∞ for two drug components. The statistical outcome for bioequivalence was shown in table 3. Cohort 1 study reveals that the ratios are about 96.7-99.7 and 87.7-92.8 respectively for Sitagliptin and Metformin components. Cohort 2 study reveals that the ratios are about 90-98.6 and 97-98.7 respectively for Sitagliptin and Metformin components. The outcome of bioequivalence was indicative and the criteria of bioequivalence was satisfied for Cohort 2. The intra-subject variability (Cohort 1) for Metformin component utilizing the ln-transformed data of the AUCt and CMax were about 22%. The lower confidence limits for Metformin (Cohort 1) was below 80% and can be improved with increase of sample size.
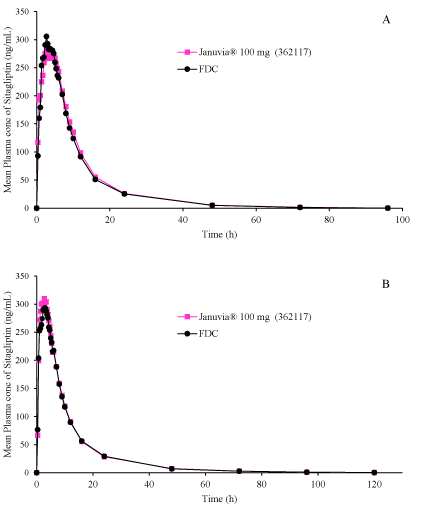 Figure 1: Mean plasma profiles of Sitagliptin from Januvia®100 mg and FDC (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged Release Tablets) under fasted state (A) and fed state (B).
Figure 1: Mean plasma profiles of Sitagliptin from Januvia®100 mg and FDC (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged Release Tablets) under fasted state (A) and fed state (B).
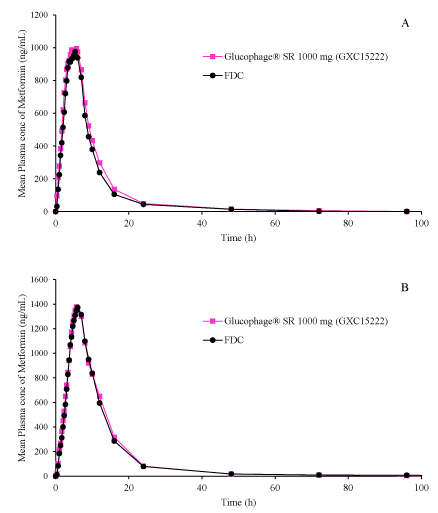 Figure 2: Mean plasma profiles of Metformin from Glucophage®1000 mg and FDC (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged Release Tablets) under fasted state (A) and fed state (B).
Figure 2: Mean plasma profiles of Metformin from Glucophage®1000 mg and FDC (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged Release Tablets) under fasted state (A) and fed state (B).
| Table2: Scheme of clinical study. | ||||
| Period | Cohort-1 (n = 12) | Cohort-2 (n = 12) | ||
| Group-1 (n = 6) | Group-2 (n = 6) | Group-1 (n = 6) | Group-2 (n = 6) | |
| 1 | Fixed Dose Combination* | Januvia® 100 mg and Glucophage® SR 1000 mg | Fixed Dose Combination* | Januvia® 100 mg and Glucophage® SR 1000 mg |
| 2 | Januvia® 100 mg and Glucophage® SR 1000 mg | Fixed Dose Combination* | Januvia® 100 mg and Glucophage® SR 1000 mg | Fixed Dose Combination* |
| *Fixed dose combination comprising of Sitagliptin 100 mg and Metformin hydrochloride 1000 mg and is manufactured by AET Laboratories Pvt Ltd., India. | ||||
| Table 3: Bioequivalence results. | ||||||
| Parameter | Sitagliptin | Metformin | ||||
| Ratio (T/R) | LCL at 90% | UCL at 90% | Ratio (T/R) | LCL at 90% | UCL at 90% | |
| Cohort 1 | ||||||
| CMax | 99.732 | 83.24 | 119.50 | 92.752 | 79.10 | 108.76 |
| AUCt | 96.688 | 91.32 | 102.37 | 88.464 | 75.56 | 103.57 |
| AUC∞ | 96.658 | 91.36 | 102.27 | 87.671 | 75.38 | 101.97 |
| Cohort 2 | ||||||
| CMax | 89.981 | 84.64 | 95.66 | 98.734 | 87.84 | 110.99 |
| AUCt | 98.537 | 96.77 | 100.34 | 96.985 | 91.06 | 103.30 |
| AUC∞ | 98.555 | 96.70 | 100.45 | 98.011 | 91.09 | 105.46 |
Safety and tolerability
A total of eight mild adverse event (AEs) were reported in five subjects (Table 4). None of these AEs were classified as serious AEs and the volunteers completely recovered from all AEs before the end of the study. From the total of subjects (5 subjects) having experienced AEs, 40% experienced AEs after treatment with FDC (2 subjects) and 100% experienced adverse events (AEs) after treatment with reference formulations (5 subjects).
| Table 4: Reported adverse events# | |||||||||
| Adverse Events | Mild | Moderate | Severe | Total | Total | ||||
| Related | NR* | Related | NR* | Related | NR* | Related | NR* | R + NR | |
| Study treatment (n = 24): Fixed dose combination-Nervous system disorders | |||||||||
| Headache MedDRA Code 100192111 |
2 (8.33%) No **04, 05 |
2 (8.33%) | 2 (8.33%) | ||||||
| Study treatment (n = 24): Reference-Nervous system disorders | |||||||||
| Headache MedDRA Code 100192111 |
4 (16.67%) No **04, 05, 09, 20 |
4 (16.67%) | 4 (16.67%) | ||||||
| Study treatment (n = 24): Reference-Gastrointestinal disorders | |||||||||
| Nausea MedDRA Code 10028813 |
2 (8.33%) No**05, 21 |
2 (8.33%) | 2 (8.33%) | ||||||
| *No Reasonable possibility; Related=Reasonable possibility; **Subject identification number; #: The overall sum of the percentages presented is 140% instead of 100.0% because there were two subjects who experienced AEs after both test and reference. | |||||||||
Discussion
Present bioequivalence study was conducted to show bioequivalence between Sitagliptin 100 mg and Metformin hydrochloride 1000 mg prolonged-release tablets of AET laboratories Pvt. Ltd. (test) and Januvia® 100 mg Filmtabletten of Merck Sharp & Dohme Ltd., United Kingdom co-administered with Glucophage® SR 1000 mg Prolonged-release Tablets of Merck Sharp & Dohme Ltd., United Kingdom (reference) in healthy volunteers. A total of 24 healthy volunteers (nine male subjects and fifteen female subjects) were enrolled in the study. As per regulatory guidelines [9], the number of evaluable subjects in a bioequivalence study should not be less than 12. Hence, 12 subjects per cohort were considered adequate considering the aim of the pilot study, which was to get the initial estimate of the comparative bioavailability of the treatments compared.
The results demonstrate that the FDC was bioequivalent for both components in Cohort 2 study and for Sitagliptin in Cohort 1 study; while the outcome was indicative for Metformin component in Cohort 1 study. It was evident that Metformin showed an intra-subject variability of about 22% in Cohort 1 study. Therefore it could be possible for Metformin to show bioequivalence with adequate sample size of approximately 38-60 evaluable subjects for demonstrating bioequivalence, considering the assumed Test/Reference Ratios of 90-110%, with a power of 80-90%.
Further, the FDC and individual reference formulations showed mild AEs and were similar to the reported adverse events [10]. In fact the, percentage of AEs for FDC was low compared to the individual reference formulations (Table 4). The results reveal that there were no safety issues with the formulations tested in the bioequivalence study. For the reference product a total of 6 adverse events were reported in 5 subjects. Out of these, there were 4 incidences of head-ache and 2 incidences of nausea. For the test product, a total of 2 adverse events were reported in 2 subjects, both of which were headache. As can be seen, the incidences of adverse events for FDC were less as compared to the reference product (mono-component product). Further, the nature of the adverse event as observed in the test product correlate with those in the reference product, (for the headache parameter), while the overall incidence was less as compared to the reference product.
From in vivo behavior and safety perspective, the FDC product comprising of Sitagliptin 100 mg and Metformin hydrochloride prolonged release 1000 mg showed more or less similar in vivo behavior as it was evidenced from the bioequivalence results and safety to that of individual reference formulations. Therefore, the use one single product in the form of FDC can enhance the patient compliance.
Conclusions
The FDC (Sitagliptin 100 mg and Metformin Hydrochloride 1000 mg Prolonged release tablets) manufactured by AET Laboratories Pvt Ltd., India is showing a comparable in vivo behavior under fed state for both the drugs in comparison to Januvia® 100 mg Filmtabletten and Glucophage® SR 1000 mg Prolonged-release tablets (both medicinal products manufactured by Merck Sharp & Dohme Ltd., UK). Fasting state bioequivalence data was satisfactory for Sitagliptin component and indicative for Metformin component. The FDC is well tolerated without any serious AEs and the AEs of FDC was similar to the AEs of reference products, hence FDC can be deemed to be safe to use in human subjects.
References
- Pathak R, Bridgeman MB. Dipeptidyl Peptidase-4 (DPP-4) Inhibitors in the management of diabetes. Pharm Therap. 2010; 35: 509-513. https://goo.gl/VtqFVR
- Januvia: Targets root problems of type 2 diabetes. 2018. https://goo.gl/QK85pu
- Tarantola E, Bertone V, Milanesi G, Capelli E, Ferrigno A, Neri D, et al. Dipeptidylpeptidase-IV, a key enzyme for the degradation of incretins and neuropeptides: activity and expression in the liver of lean and obese rats. Eur J Histochem. 2012; 56: e41. https://goo.gl/EUg3a7
- Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, et al. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther. 2005; 78: 675-688. https://goo.gl/QhVSmR
- American Diabetes Association. Standards of medical care in diabetes-2014. Diabetes Care. 2014; 37: S14-S80. https://goo.gl/XPFBWt
- Glucophage SR 500 mg, 750 mg and 1000 mg prolonged release tablets. 2017. https://goo.gl/Q867TJ
- Shahid Sarwar, Delowar Hossain. Fixed dose combination and disease management. Int Res J Pharm. 2012; 3: 17-21. https://goo.gl/8oeMBs
- ICH harmonized guidelines. Integrated addendum to ICH E6 (R1): guideline For Good Clinical Practice E6 (R2). 2016. https://goo.gl/tBtC6P
- Guideline on the investigation of bioequivalence. European Medicine Agency Guidelines. 2010. https://goo.gl/99CgD7
- Subbarayan S, Kipnes M. Sitagliptin: a review. Expert Opin Pharmacother. 2011; 12: 1613-1622. https://goo.gl/yHZVM3
Authors submit all Proposals and manuscripts via Electronic Form!


























