

Case Report
Can we Diagnose Juvenile Polyposis Syndrome without Genetics? - Case Report and Review of Literature
Virtut Velmishi1*, Olta Meta1, Gentiana Cekodhima2 and Paskal Cullufi1
1Service of pediatric gastroenterology, Mother Teresa Hospital, Tirana, Albania
2Service of histopathology, Mother Teresa Hospital-Tirana, Albania
*Address for Correspondence: Virtut Velmishi, Dibra Street Nr 372, Mother Teresa Hospital Tirana, Albania; E-mail: tutimodh@yahoo.com
Dates: Submitted: 06 June 2017; Approved: 30 June 2017; Published: 04 July 2017
Citation this article: Velmishi V, Meta O, Cekodhima G, Cullufi P. Can we Diagnose Juvenile Polyposis Syndrome without Genetics? - Case Report and Review of Literature. Int J Hepatol Gastroenterol. 2017;3(1): 013-016.
Copyright: © 2017 Velmishi V, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: JPS: Juvenile Polyposis Syndrome; FAP: Familial Adenomatous Polyposis; HPMS: Hereditary Mixed Polyposis Syndrome; SPS: Serrated Polyposis Syndrome; MAP: mutY-Associated Polyposis; GS: Gorlin Syndrome; PJS: Peutz- Jeghers Syndrome
Abstract
Our case report presents a ten-year-old boy who complains abdominal pain and rectal bleeding for more than six months. Other clinical findings were digital hippocratism and failure to thrive. Colonoscopy had shown 12 polyps from anus to cecum whose histology confirmed Juvenile character, non adenomatous and free of dysplasia. Diagnosis of Juvenile Polyposis Syndrome was more probable but a differential diagnosis from other intestinal polyposis especially from hamartomatous syndromes is crucial.
Introduction
Juvenile polyposis syndrome (JPS) is a rare autosomal-dominant disorder associated with an increased risk of colorectal cancer (68% by age 60 years) and upper gastrointestinal tract cancer (10%) [1-2]. Juvenile polyps are histological distinct and consist of dilated cysts filled with mucin and lined by Stromal cells. The prevalence of JPS is estimated 1:100000. The number of polyps required to establish the diagnosis remains controversial, but most agree that patients with between three and five Juvenile polyps or any number of Juvenile polyps found in a patient with a family history of JPS should undergo evaluation [3]. The polyps in JPS are located throughout the gastrointestinal tract but predominate in the colon. These usually begin appearing before age 20, but the term Juvenile refers to the type of polyp, not to the age of patient [4]. In children, the most common clinical manifestation is colorectal bleeding, occurring at an average age of 9 years. The blood loss may be occult with development of iron deficiency anemia or overt gastrointestinal bleeding [5]. In addition, diarrhea and protein losing enteropathy have been reported. A significant number of children with JPS have been found to have other abnormalities, including digital clubbing, macrocephaly, alopecia, cleft lip or palate, congenital heart disease, genitourinary tract abnormalities, and mental retardation.
Case Presentation
We would like to present the case of a ten- year -old boy complaining abdominal pain and rectal bleeding for the last 6 months. Currently, his mother has noted something to pass out of anal merge after defecation. Initially, we thought for rectal prolaps. This boy presented also digital hippocratism (Figure 1) and failure to thrive (Weight = 19 kg; height = 118.5 cm). We had a suspicion for cystic fibrosis but sweat test resulted normal. In objective examination this boy had not facial dysmorphy, no heart murmurs, lungs were clear and abdomen was slightly meteoric. His father referred also rectal bleeding time by time but he was not been examinated.
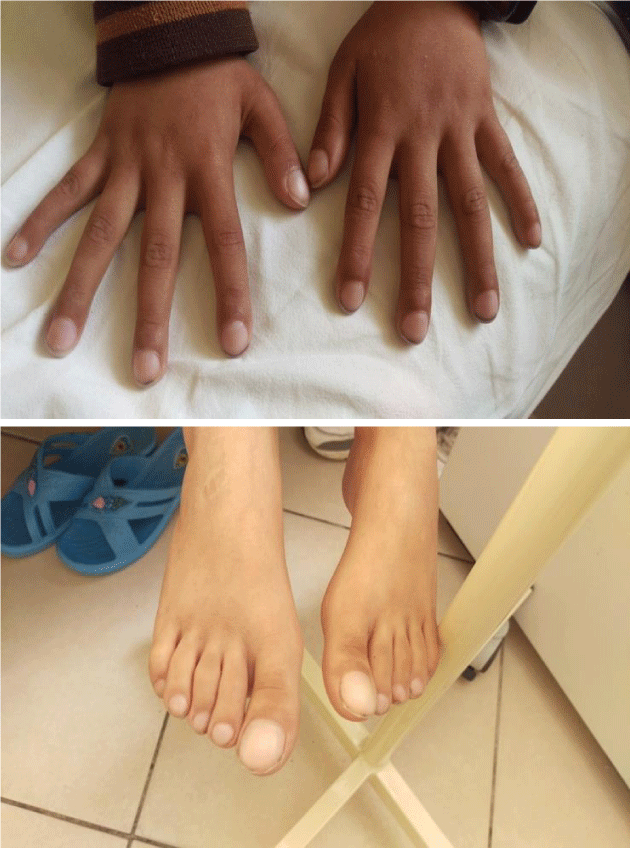 Figure 1: Digital hippocratism.
Figure 1: Digital hippocratism.
The first laboratory test had shown a Hb = 7,4 mg/ dl , hypoalbuminemia 2,9 mg/ dl. The rest of laboratory tests were normal. Heart Ultrasonography and EKG resulted normal. Abdominal ultrasound did not reveal anything except meteorism. Celiac antibodies were between the normal ranges. In this context we decided to perform a colonoscopy. During examination, we have seen 12 polyps from anus to the cecum. The most part of them was in the rectum and colon descendens. Characteristic finding was the long peduncule (Figure 2). We have performed polypectomy sending some of them to histopathology service. The response of pathology showed a benign polyp with cystic structure characteristic of solitary Juvenile polyps. Hematoxylin eosin stain of a polyp showed spherical surface epithelium with cystically dilated glands in lamina propria (Figure 3).
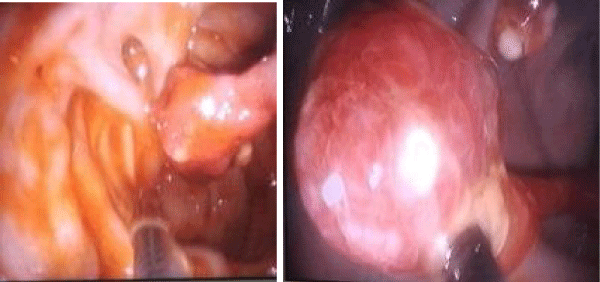 Figure 2: A pedunculate polyp in cecum (left) and polypectomy (right).
Figure 2: A pedunculate polyp in cecum (left) and polypectomy (right).
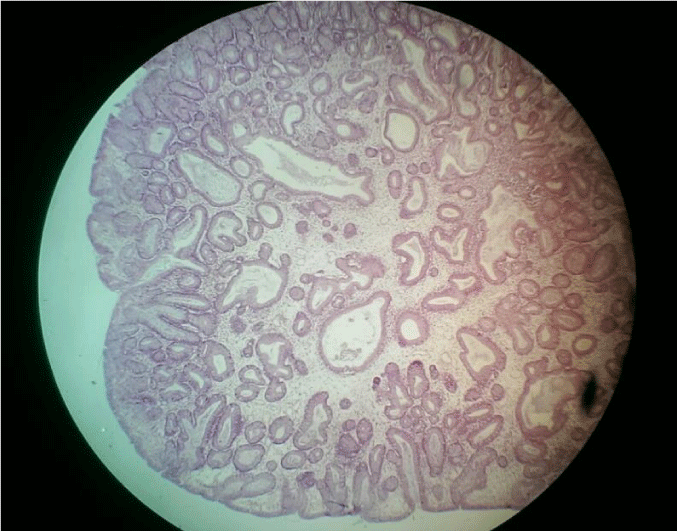 Figure 3: Hematoxylin eosin stain of a juvenile polyp which shows spherical surface epithelium with cystically dilated glands in lamina propria).
Figure 3: Hematoxylin eosin stain of a juvenile polyp which shows spherical surface epithelium with cystically dilated glands in lamina propria).
At the same time, we performed also an upper digestive endoscopy but the image was compatible with a severe nodular gastritis. We did not find any polyp in stomach or duodenum. We took also some samples for biopsy which has shown helicobacter pylori gastritis. We have started triple therapy with omeprasole, amoxycilline and clarithromicine. After a month we have performed another colonoscopy cutting the rest of colonic polyps. In the same time this boy underwent to abdominal and head CT which did not reveal any abnormality. We have followed this boy for more than a year .He had less episodes of rectal bleeding and the last level of hemoglobin was 10.8 g/ dl.
Discussion
Juvenile polyposis syndrome may be caused by mutations in two genes: SMAD4/DPC4 located on chromosome 18q21 and BMPR1A on chromosome 10q21-22 [5,6]. The indication for performing genetic testing for JPS is detection of three to five Juvenile polyps in a patient, detection of a Juvenile polyp and a family history of JPS, or being a parent or sibling of a known mutation carrier. Testing involves DNA sequencing for mutations in SMAD4 and BMPR1A, which are associated with JPS in approximately 35 to 50% of patients with JPS [7]. Performing genetic test in our daily practice is hard because of high cost. Sometimes, we are constraint to be based on clinical and histological data. First of all we have to differentiate syndromes with intestinal polyposis.
Intestinal polyposis can be divided, based on histology into the broad categories of Familial Adenomatous Polyposis (FAP); Hamartomatous Polyposis Syndromes, and other rare polyposis syndromes such as Hereditary Mixed Polyposis Syndrome (HPMS) and Serrated Polyposis Syndrome (SPS). Clinical features or associated abnormalities are also very important for a differential diagnosis. For example in Cowden syndrome are characteristic facial trichilemmomas, nontoxic multinodular goiter, thyroid, and breast cancer, whereas in Bannayan-Riley-Ruvalcaba syndrome predominates symptoms such as macrocephaly, hyperpigmentation of the genitalia and retarded psychomotor development. Comparing those syndromes with our case clinical data are sufficient to exclude JPS
Peuts-Jeghers Syndrome is an autosomal dominant inherit disorder characterized by intestinal hamartomatous polyps but the most significant difference from our case is a distinct pattern of skin and mucosal melanin deposition which is not found in JPS. Patients with Peutz-Jeghers syndrome have a 15-fold increased risk of developing intestinal cancer compared with the general population [8-13].
There are some other colonic polyposis syndromes in children like FAP (Familial Adenomatous Polyposis), AFAP (Attenuated Familial Adenomatous Polyposis) and MAP (mutY –Associated Polyposis). Several specified variants of FAP, namely Gardner syndrome and Turcot syndrome have been identified. Individuals with Gardner syndrome develop adenomatous polyps throughout the GI tract, accompanied by extracolonic manifestations, including periampullary adenomas, papillary carcinoma of the thyroid, hepatoblastoma, osteoma of the mandible and skull, epidermal cysts, and desmoids tumors. Turcot syndrome, another variant of FAP, is a rare autosomal recessive disorder that can present with brain tumors (glioblastoma multiforme, medulloblastoma) and colonic adenomas that frequently become malignant in those younger than 30 years. It was initially described by Turcot [14] and again in 1969 by Baughman, et al. [15] .The most important finding in those syndromes is polyp histology which shows data compatible with adenoma differentiating from histology of JPS which is more compatible with hamartoma.
As we mentioned above JPS took place between hamartomatous polyposis syndromes which encompasses several syndromes, mainly Peutz - Jeghers Syndrome, PTEN - associated hamartomatous syndromes including Cowden syndrome, Bannayan – Riley - Ruvalcaba syndrome and Cronkhite Canada syndrome. The last syndrome is described in 1955, presented at average age 59 years, accompanied with multiples intestinal polyps and ectoderm anomalies, including hyperpigmentation of the skin, alopecia, and onychoheterotopia [16]. Even Cronkhit Canada syndrome can be differentiated easily by JPS because of other abnormalities and presentation time despite a high similarity in histology of polyps.
HPMS is extremely rare. It is characterized by familial presentation of colorectal polyps that have mixed histologic elements with both edematous and hyperplastic features [17]. In 1960, Gorlin and Goltz [18] initially described Gorlin Syndrome (GS), also termed nevoid basal cell carcinoma syndrome. Herzbers and Wiskemann further associated GS with medulloblastoma in 1963. GS commonly presents with hamartomatous gastric polyps, palmar pits, short metacarpals, odontogenic keratocytes, intracranial calcifications, skeletal malformation and neoplasia. First defined in 2000, Burt and Jass [19] described SPS, which is characterized by multiple, large serrated polyps within the colon leading to a high risk of colorectal cancer. Although its inheritance is unclear, hereditary and sporadic cases have been described in the literature. The basic of differential diagnosis between HPMS and JPS remains histology features.
The World Health Organization criteria for diagnosis of JPS are one or either:
1. More than 5 Juvenile polyps in the colon or rectum or
2. Juvenile polyps throughout the gastrointestinal tract or
3. Any number of Juvenile polyps in a person with a family story of Juvenile polyposis [20]
In our case, we found 12 polyps compatible with Juvenile type in histology. Our boy had not other abnormality despite finger clubbing and failure to thrive. Lab test showed anemia and hypoalbuminemia. Father had rectal bleeding but he did not accept to perform digestive endoscopy. According to WHO criteria it is more easily to diagnose JPS without genetics but based only on histology and few clinical findings we may mistake the diagnosis with other hamartomatous polyposis syndromes. A better knowledge of intestinal polyposis associated with a good evaluation of clinical data and histology can be crucial to diagnose JPS.
Conclusion
Molecular genetics diagnosis may be gold standard not only for basic diagnosis but also for a better prediction of cancer risk in children and adolescent. When the possibilities to perform genetic analysis are scarce we recommend combining a clinical, endoscopic and histological data for a basic diagnosis of Juvenile polyposis syndrome. As a conclusion, we can say that JPS could be diagnosed without genetics.
References
- Brosens LAA, Langeveld D, Van Hattem WA, Giardello F.M and Offerhaus GJA. Juvenile polyposis syndrome. World J Gastroenterol. 2011; 17: 4839-4939-4844. https://goo.gl/V5m23A
- Watanabe A, Nagashima H, Motoi M and Ogawa K. Familial Juvenile polyposis of the stomach. Gastroenterology. 1979; 77; 148-151. https://goo.gl/jn4vis
- Jass JR, Williams CB, Bussey HJ and Morson BC. Juvenile polyposis-a precancerous condition. Histopathology. 1988; 13: 619-303. https://goo.gl/7Z4QZv
- Brosens LA, VAN Hatten WA, Jansen M, DE Leng WW, Giardello FM and Offerhaus GJ. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007; 7: 29-46. https://goo.gl/hZvvmj
- Schreibman IR, Baker M, Amos C, Mc Garryty TJ. The hamartomatous polyposis syndromes: A clinical and molecular review. Am J Gastroenterol. 2005; 100: 476-90. https://goo.gl/nNbSTb
- Howe JR, Sayed MG, Ahmed AF, Ringold J, Larsen-Haidle J, Merg A, et al. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, AND ACVR1 mutations. J Med Genet. 2004; 41: 484-91. https://goo.gl/EFU4N6
- Gene Tests. Version current at November 9, 2006: http// GENETests.org
- Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987; 316: 1511-4. https://goo.gl/Uh5syR
- Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008; 88: 779-817. https://goo.gl/NUJVQt
- Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000; 119: 1447-53. https://goo.gl/4CFmUT
- Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998; 128: 896-9. https://goo.gl/KH5edb
- Trau H, Schewach-Millet M, Fisher BK, Tsur H. Peutz-Jeghers syndrome and bilateral breast carcinoma. Cancer. 1982; 50: 788-92. https://goo.gl/6s4tVP
- Gruber SB, Entius MM, Petersen GM, Laken SJ, Longo PA, Boyer R, et al. Pathogenesis of adenocarcinoma in Peutz-Jeghers syndrome. Cancer Res. 1998; 58: 5267-70. https://goo.gl/1d3vRJ
- Turcot J, Despres JP, St Pierre F. Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum. 1959; 2: 465-8. https://goo.gl/FcPBCn
- Baughman FA, List CF, Williams JR, et al. The glioma-polyposis syndrome. N Engl J Med. 1969; 281: 1345-46. https://goo.gl/MSD1vM
- Slavik T, Montgomery EA. Cronkhite–Canada syndrome six decades on: the many faces of an enigmatic disease. J Clin Pathol. 2014; 67: 891-7. https://goo.gl/SqMFxD
- Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin North Am. 2008; 88: 779-817. https://goo.gl/MwS54u
- Gorlin RJ. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore). 1987; 66: 98-113. https://goo.gl/Thsz5d
- Guarinos C, Sánchez Fortun C, Rodríguez Soler M, Alenda C, Paya A, Jover R. Serrated polyposis syndrome: molecular, pathological and clinical aspects. World J Gastroenterol. 2012; 18: 2452-61. https://goo.gl/SqvxdE
- Stoler Mark A, Mills Stacey E, Carter Darryl, Joel K Greenson, Reuter Victor E. Sternberg's Diagnostic Surgical Pathology. Hagerstwon, MD: Lippincott Williams & Wilkins. 2009. https://goo.gl/VwKS2Q
Authors submit all Proposals and manuscripts via Electronic Form!


























