

Research Article
A Novel Mechanism to Reduce Myocardial Remodeling: The Nitric Oxide Synthase Inhibitor L-NAME Shields the Remote Myocardium from Apoptosis and Fibrosis after Myocardial Infarction in-vivo
Felix M. Heidrich, Anna Ritzkat, David M. Poitz, Annette Ebner, Melissa M. Cremers, Tobias F. Ruf, Silvio Quick, Christian Pfluecke, Ruth H. Strasserand Stephan Wiedemann
Felix M. Heidrich*, Anna Ritzkat, David M. Poitz, Annette Ebner, Melissa M. Cremers, Tobias F. Ruf, Silvio Quick, Christian Pfluecke, Ruth H. Strasserand Stephan Wiedemann
Technische Universitat Dresden, Heart Center Dresden University Hospital, Dresden, Germany
*Address for Correspondence: Felix M. Heidrich, Department of Cardiology, Technical University Dresden, Heart Center Dresden University Hospital, Fetscherstr, 76, 01307 Dresden, Germany
Dates: Submitted: 22 December 2016; Approved: 10 February 2017; Published: 13 February 2017
Citation this article: Heidrich FM, Ritzkat A, Poitz DM, Ebner A, Cremers MM, et al. A Novel Mechanism to Reduce Myocardial Remodeling: The Nitric Oxide Synthase Inhibitor L-NAME Shields the Remote Myocardium from Apoptosis and Fibrosis after Myocardial Infarction in-vivo. Int J Clin Cardiol Res. 2017;1(1): 020-030
Copyright: © 2017 Heidrich FM, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Experimental myocardial infarction; Myocardial remodeling; Apoptosis; Endothelial nitric oxide synthase; Nitric oxide
Abstract
Rationale: Following Myocardial Infarction (MI), apoptosis contributes to the processes of myocardial remodeling. Nitrosative stress is a well-known inductor of myocardial apoptosis. Activation and differential phosphorylation of Endothelial Nitric Oxide Synthase (eNOS) may increase its uncoupling potential, resulting in an excess of peroxynitrite formation. However, the pathophysiologic impact of eNOS signaling in the remote myocardium after MI until now is unexplored.
Objective: The goal of the present study was to assess the impact of eNOS signaling on Heart Failure (HF) development, the induction and execution of apoptosis, and fibrotic remodeling in the remote, non-ischemic myocardium in vivo.
Methods and Results: NOS inhibition was achieved in a rat MI animal model in vivo using the specific NOS-inhibitor N?-Nitro-L-arginine methyl ester (L-NAME). L-NAME treatment for 14 days after MI significantly ameliorated para-clinical and clinical HF indices (N-terminal pro-atrial natriuretic peptide, heart-to-body weight ratio, lung water content). Moreover, L-NAME treatment prevented effector caspase 3 activation and led to significantly less apoptosis execution and fibrotic remodeling in the remote myocardium. Notably, L-NAME treatment prevented harmful eNOS-phosphorylation at threonine (495), resulting in a preserved serine (1177)/threonine (495) eNOS phosphorylation ratio.
Conclusion: These data demonstrate that apoptosis execution and fibrotic remodeling after MI in the remote, non-ischemic myocardium are the consequence of increased eNOS-phosphorylation at threonine (495).Treatment with L-NAME not only restored eNOS serine (1177)/threonine (495) phosphorylation ratio but also reduced apoptosis execution and fibrotic remodeling. Therefore, modulating differential eNOS phosphorylation might become an important pharmacological target for the development of novel tailored therapies to prevent myocardial remodeling and the development of HF.
Introduction
Despite treatment advances, cardiovascular diseases and particularly acute coronary syndrome that might lead to Heart Failure (HF) still is a leading cause of morbidity and mortality worldwide [1,2]. In the early phase following Myocardial Infarction (MI), acute ischemic symptoms prevail whereas the loss of viable myocardium may cause progressive Left Ventricular (LV) dysfunction and clinically the development of symptomatic HF. From a pathophysiologic perspective, the central underlying process contributing to the development of HF is ventricular remodeling, which involves the key processes of apoptosis, necrosis und hypertrophy [3-5]. While it is acknowledged that the development of HF is the result of progressive LV dysfunction after acute MI, the molecular mechanisms promoting hypertrophy, apoptosis, necrosis and fibrosis are still not completely understood.
Although the loss of viable, contractile myocardium via apoptosis and necrosis can be in part compensated by hypertrophy, scar healing usually cannot be completely prevented. The consequence is progressive HF with reduced LV Ejection Fraction (LVEF), which results in systemic compensatory mechanisms, namely activation of the renin-angiotensin-aldosterone-system [6]. However, these initially compensatory effects on hemodynamics nourish the mid- and long-term continuous negative adaptive remodeling processes of the heart itself, further promoting the progression of LV dysfunction (circulus vitiosus).
Apoptosis plays a crucial role in post MI ventricular remodeling [7,8]. While it has been studied extensively in the infarct- and border zones after MI, the emphasis of apoptosis in the non-infarcted remote myocardium is less pronounced and is still disputed [9]. Nevertheless, when the total apoptotic myocardial cellular population after MI is considered, apoptosis in the remote myocardium can reach up to 70%of all myocardial apoptotic cells at 12 weeks after MI [10]. Induction of apoptosis is multifactorial with neuro-humoral, mechanical and notably-oxidative and nitrosative stress being among the most important trigger factors [11,12].
A well-known and potent inductor of apoptosis is increased nitrosative stress [13]. Nitric Oxide (NO) synthases (NOSs) are commonly believed to exhibit cardioprotective effects during and after MI due to NO production, given the availability of the essential NOS cofactors including tetrahydrobiopterin (BH4) [14-17]. The Endothelial NOS (eNOS) is the predominant cardiac isoform and is detectable in endothelial cells and cardiomyocytes [18]. Phosphorylation increases eNOS activity with serine (1177) being one of the most important phosphorylation sites for increasing NO production [19]. However, it is of note that an improper electron transfer can result in eNOS uncoupling, a condition when superoxide anion instead of NO is produced [15]. Various causes of eNOS uncoupling are described, including eNOS phosphorylation at threonine (495) which promotes non-stable eNOS dimerization [15,20,21]. Superoxide anions themselves do not possess pro-apoptotic potential [21]. However, they can easily react with NO to form the highly reactive peroxynitrite [22,23]. Notably, peroxynitrite not only increases nitrosative stress but also inactivates the eNOS cofactor BH4 which acts as a stabilizer of non-stable eNOS dimers, therewith further propagating the uncoupling process [13-15]. A circulus vitiosus is established.
In this study, the impact of NOS inhibition with the specific NOS-inhibitor L-NAME on the development of HF after acute MI and its impact on apoptosis induction, execution and fibrotic remodeling in the remote non-ischemic myocardium was studied in a rat MI animal model in vivo.
Material and Methods
Experimental myocardial infarction (MI), sampling, determination of infarct sizes
Left Anterior Descending coronary artery (LAD), Ligation (LIG) and sham operation (sham) in adult male Wistar rats were performed as previously described [24-27]. After the operation, anesthesia was terminated and the animals were kept alive and conscious for another 14 days with the LAD remaining occluded in LIG animals to resemble chronic MI. For post operation treatment (L-NAME vs. placebo), animals were allocated to one of four treatment groups (Refer to section 2.2). After 14 days, animals were re-anesthetized and euthanized and transmural drill biopsies of the posterior non-infarcted wall of the Left Ventricle (LV) taken (n = 10-11 per group). Hearts from another 6-8 animals per treatment group were kept for further processing for light microscopy and immunofluorescence experiments. Another 4 animals per treatment group were used for preliminary determination of infarct sizes as previously described by means of a Langendorff perfusion setup using propidium iodide and fluorescent microspheres to visualize the infarcted area, the area at risk and viable myocardium [28]; representative images are shown in the Appendix A (Figure 1A). All animal procedures were performed in agreement with the "Guide for the Care and Use of Laboratory Animals" [29] and were approved by the local committee (#24-9168.11-1/ 2012-53).
Treatment groups
Adult male Wistar rats were allocated to one of four treatment groups according to the performed operation (LIG vs. sham) and post interventional treatment (L-NAME vs. placebo), Table 1. L-NAME (50 mg/kg body weight, dissolved in 0.9% sodium chloride) or placebo (i.e., sodium chloride, 0.9% sodium chloride in 100 ml water) were administered orally and available ad libitum immediately after animals awoke from anesthesia for a period of 14 days. All animals showed a comparable drinking behavior.
Echocardiographic assessment of systolic heart function and determination of heart failure (HF) development
To estimate Left Ventricular Ejection Fraction (LVEF), transthoracic echocardiography was performed prior to operation and 14 days later prior to euthanasia in each animal. LVEF was estimated using the monoplane Simpson's method from 2D-echocardiography recordings according to the following equation: LVEF = (EDVol-ESVol)/ EDVol x 100 [30]. HF development was assessed via serum N-terminal pro-atrial natriuretic peptide (NT - proANP) determination by Enzyme-Linked Immunosorbent Assay (ELISA) and determination of the heart-to-body weight ratios as previously described [24-26]. Additionally, lung water content was estimated from the difference of lung wet weight to lung dry weight as previously described [31].
Protein determination
Protein determination was performed according to the method of Bradford, et al. [24-27] as previously described.
Tissue preparation, immunoblot analysis
Heart biopsies were homogenized in buffer A (50 mM TrisHCl, 5 mM EDTA, 2 mM EGTA, 1 mM PMSF, 1 mM benzamidine) using a Polytron (LS 10-35, Kinematica, Luzern, Switzerland) (3 6 sec, 14.000 rpm, 4 C). The homogenate was centrifugated (360 g, 10 min, 4 C). The resulting supernatant was again centrifugated at 50.000 g (45 min., 4 C) to separate the soluble fraction from the particulate fraction, leaving the cytosol as the supernatant. Aliquots were taken for western blotting. Samples containing 30 g of cytosolic protein were separated on 12% SDS-polyacrylamide gels according to the method of Laemmli, et al. [32] and transferred to Polyvinylidene Fluoride (PVDF) membranes using the method of Towbin, et al. [33], as reported previously [24,27]. Pre-stained molecular weight standards were electrophoresed and transferred in parallel. Primary antibodies targeting -actin, bcl-2, bax, eNOS, p-eNOS (serine 1177), p-eNOS (threonine 495), caspase 3 and 8, cleaved caspase 3 and 8 (Cell Signaling, Danvers, USA; Calbiochem, Schwalbach, GER; Santa Cruz, Heidelberg, GER; as detailed in (Table 1A)) were detected using monoclonal goat-anti-rabbit or sheep-anti-mouse IgG HRP-linked secondary antibodies (all Cell Signaling, Danvers, USA; as detailed in (Table 1A)). The PVDF membranes were blocked with 5% milk and 0.1% Tween-20 in PBS ("blocking buffer") for 1 hour, followed by incubation with antibodies diluted in blocking buffer at their respective dilutions over night at 4 C. Incubation with secondary antibodies was performed for 1 hour in blocking buffer at room temperature. The detection of the specific protein bands was performed using Western Lightning ECL chemiluminescence kit (Perkin Elmer). For semi-quantitative analysis, intensities of protein bands were quantified by densitometry using Quantity One software (BioRad, Hercules, USA) and normalized to the respective inactive or non-phosphorylated protein (caspase 8, eNOS) or -actin, as reported previously [34]. The protein ratios of cleaved caspase 8/pro-caspase 8, p-eNOS (threonine 495)/eNOS and p-eNOS (Ser 1177)/p-eNOS (threonine 495) were determined sequentially from one single membrane each after protein-stripping. Each sample was analyzed in three independent repetitions. Sample normalization to compare target protein expression levels between different samples on the same blot or across several blots was performed with a loading control to normalize the data. In preliminary experiments, target protein aliquots were analyzed in the presence of excess antigenic peptide. The peptides fully blocked staining of the specific bands. Increased amounts of protein were analyzed on the gels to ensure the linearity of the analysis in the protein range used.
Cryosections, light microscopy, detection of apoptosis (M30, TUNEL)
Cryosections were prepared as previously described [25,27]. A total of three short-axis section levels per animal and heart were used for data acquisition and quantifications. Total collagen content was estimated with picrosirius red staining as previously described [24]. Apoptotic cells were detected by an M30 antibody, which recognizes caspase 3 cleaved ceratin-18 neo-epitope (M30 CytoDeathTM, Enzo Life Sciences, Lorrach, GER). ACy-3 conjugated anti-mouse antibody (CyTM, Jackson, UK) was used as the secondary antibody. Counterstaining was done with 4', 6-Diamidin-2-phenylindol (DAPI) for 3 minutes (0,5 g/ml in PBS; Applichem, Darmstadt, GER). Final fixation was performed using Mount Flour (BroCyl, Luckenwalde, GER). Terminal Deoxy Nucleotidyl Transferase (TdT)-mediated dUTP nick end-labeling (TUNEL) was performed as previously described [25,26] using the "in situ cell death detection kit" (Roche, Mannheim, GER), and counterstaining with DAPI and final fixation were done as described above. Samples were viewed with an inverted fluorescence microscope (Axio Vert S100, Zeiss, Gottingen, GER) at a 10- to 40- fold magnification. Image acquisition was done using the Spot RT KE Slider camera (SPOT Imaging, Sterling Heights, USA). For quantification, 3 visual fields of pre-specified areas of the remote LV myocardium from the three pre-specified short-axis section levels were analyzed using an ImageJ software. Data was recorded as the relative red-channel content (in the case of picrosirius red staining) or the number of apoptotic cells in relation to the total cell count per visual field and averaged among all available visual fields and section levels.
Figure 1
Left ventricular systolic function and onset of heart failure.A: LIG treatment significantly reduced LVEF. L-NAME treatment did not improve LVEF in LIG animals.
B-D: Evaluation of heart failure development.
B: LIG treatment caused a significant increase in NT-proANP serum levels. No significant increase of NT-proANP serum levels was obvious in post operation L-NAME treated LIG animals compared to sham.
C: Heart-to-body weight ratio significantly increased in LIG treated animals. This was seen significantly attenuated in post operation L-NAME treated LIG animals.
D: Lung water content was in trend increased in native LIG animals. Significantly less lung water content was observed in L-NAME treated LIG animals compared to placebo treatment.
LVEF, left ventricular ejection fraction. L-NAME, N?-Nitro-L-arginine methyl ester. NT-proANP, N-terminal pro-atrial natriuretic peptide. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending coronary artery (LAD). sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Statistical annotations: n.s. not significant; xstatistic trend (p = 0.2);*p = 0.05; **p = 0.01;***p = 0.001, n = 12-14 per group.
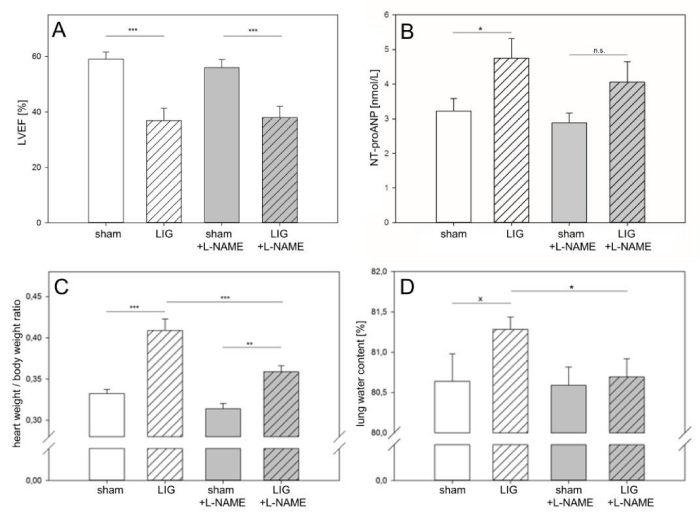
Table 1
| Table 1: Treatment groups. Post operation treatment (L-NAME or placebo) was administered orally and available ad libitum immediately after animals awoke from anesthesia for a period of 14 days. L-NAME, N?-Nitro-L-arginine methyl ester. LAD, Left Anterior Descending coronary artery. LIG, treatment group experiencing true LAD ligation. Sham, treatment group experiencing sham operation without ligation of the LAD. n. number of animals. | ||||
| Group | Abbreviation | n | Post operation treatment (for 14 days) | Operation |
| 1 | LIG | 20 | Placebo (i.e. 0.9% sodium chloride) | LAD ligation (for 14 days) |
| 2 | Sham | 18 | Placebo (i.e. 0.9% sodium chloride) | Sham |
| 3 | LIG + L-NAME | 18 | L-NAME (50 mg/kg body weight, dissolved in 0.9% sodium chloride) | LAD ligation (for 14 days) |
| 3 | sham + L-NAME | 17 | L-NAME (50 mg/kg body weight, dissolved in 0.9% sodium chloride) | sham |
Statistical Analysis
Data analysis was done in SPSS (IBM, Armonk, USA). Normality distribution of the data was confirmed using chi-squared (X2) test. Analysis of variance (ANOVA) was used in multiple comparisons with post hoc testing by means of the fisher-t-test. Additionally, student t-test was used for comparison of two groups. All values are given as means standard error of the mean (SEM). p = 0.05 was considered statistically significant and indicated as p = 0.01 indicated as and p = 0.001 indicated as p = 0.2 was defined as trend towards statistical significance and indicated as X. Graphs were prepared with Sigma Plot (Systat Software Inc., San Jose, USA).
Results
NOS inhibition by L-NAME ameliorates heart failure
Left Ventricular Ejection Fraction (LVEF) was observed to be significantly reduced in animals that underwent true Ligation (LIG) of the Left Anterior Descending Coronary Artery (LAD) compared to sham treatment (Figure 1A). Post operation treatment with L-NAME had no significant effect on LVEF in sham or LIG treated animals (Figure 1A). However, when evaluating para-clinical and clinical indices of Heart Failure (HF), amelioration of HF was observed in L-NAME treated LIG animals (Figure 1B-D). While significant NT-proANP elevation was present in placebo-treated LIG animals compared to sham treatment, NT-proANP elevation failed to reach statistical significance in post operation L-NAME treated LIG animals (Figure 1B). Heart-to-body weight ratios increased in both, placebo and L-NAME treated LIG animals. However, this increase was significantly less in L-NAME treated LIG animals compared to placebo treatment (Figure 1C). Finally, placebo-treated LIG animals showed a trend towards increased lung water content, indicative of progressive left ventricular HF. Of note, no such a trend was seen in L-NAME treated LIG animals (Figure 1D).
NOS inhibition by L-NAME prevents apoptosis inductionin the remote myocardium
LIG treatment resulted in significant activation of the extrinsic apoptotic pathway as assessed by cleaved caspase 8/total caspase 8 protein ratios in both, placebo and L-NAME treated LIG animals without statistically significant differences between the two groups (Figure 2A). No activation of the intrinsic apoptotic pathway assessed by the bcl-2/bax protein expression ratio was observed in the remote myocardium in LIG animals, regardless of post-operation treatment (Figure 2B). Notably, effector caspase 3 activation in terms of cleaved caspase 3 protein expression showed significant activation only in LIG/placebo-treated animals but did not reach statistically significant activation in LIG animals receiving L-NAME post operation (Figure 2C). Representative western blots may be viewed in the online data supplement (Figure 2A).
NOS inhibition by L-NAME reduces apoptosis execution in the remote myocardium
LIG treatment resulted in a significant increase in apoptosis execution assessed by M30 immunohistochemistry (Figure 3A + C). Of note, no statistically significant increase in apoptosis execution was observed in L-NAME treated LIG animals when compared to sham treatment (Figure 3A + C). Importantly, a significant rescue effect was obvious in L-NAME treated LIG animals when compared to placebo treated LIG animals (Figure 3A+ C). Similar effects were observed for apoptosis execution assessed with the TUNEL assay (Figure 3B + D). LIG treatment resulted in a significant increases in TUNEL positive cells whereas L-NAME treated LIG animals did not show significant increases of TUNEL positive cells when compared to sham treatment (Figure 3B + D).
NOS inhibition by L-NAME prevents fibrotic remodeling in the remote myocardium
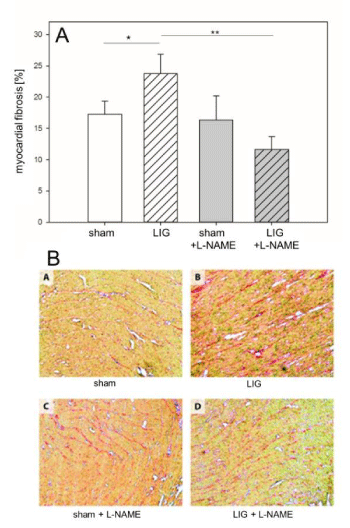
LIG treatment caused a significant increase of the total fibrotic content in the remote myocardium, assessed in cryo-sections after picrosirius red staining (Figure 4A + B). Of note, fibrotic remodeling of the remote myocardium was prevented in L-NAME treated LIG animals (Figure 4A + B).
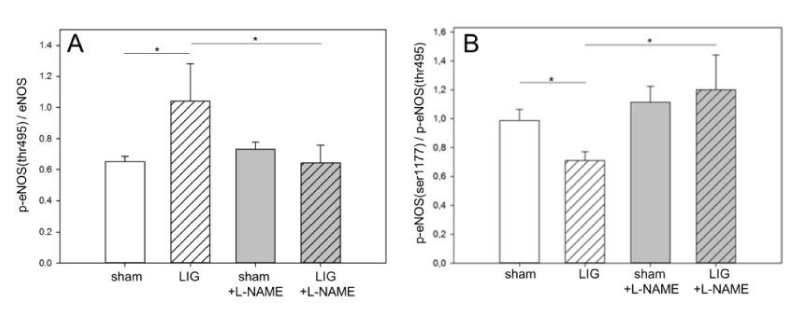
L-NAME prevents eNOS phosphorylation at threonine (495) and preserves eNOS serine (1177)/ threonine (495) phosphorylation ratio
Phosphorylation of eNOS at ser1177 and thr495 are central regulatory switches of this enzyme [15]. LIG treatment resulted in a significant increase of phosphorylation at thr495 and this translated in significantly depressed ser1177/thr495 eNOS phosphorylation ratio (Figure 5A + B). Notably, L-NAME treatment prevented excess eNOS phosphorylation at thr495 and resulted in a preserved eNOS ser1177/thr495 phosphorylation ratio (Figure 5A + B). Representative western blots may be viewed in the online data supplement (Figure 3A).
Figure 2
Remote myocardium, apoptosis induction. A: LIG treatment induced apoptosis via the extrinsic pathway as expressed in increased cleaved (active) caspase 8/pro-caspase 8 ratio. Post operation L-NAME treatment did not prevent caspase 8 activation.
B: No significant induction of apoptosis was documented assessing the intrinsic pathway, neither in native nor in L-NAME treated LIG animals.
C: Post operation L-NAME treatment prevented significant caspase 3 activation. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending coronary artery (LAD). sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Statistical annotations: n.s. not significant; xstatistic trend (p = 0.2); *p = 0.05; **p = 0.01. n=10-11 per group. Representative western blots may be viewed in the online data supplement (Figure A2).
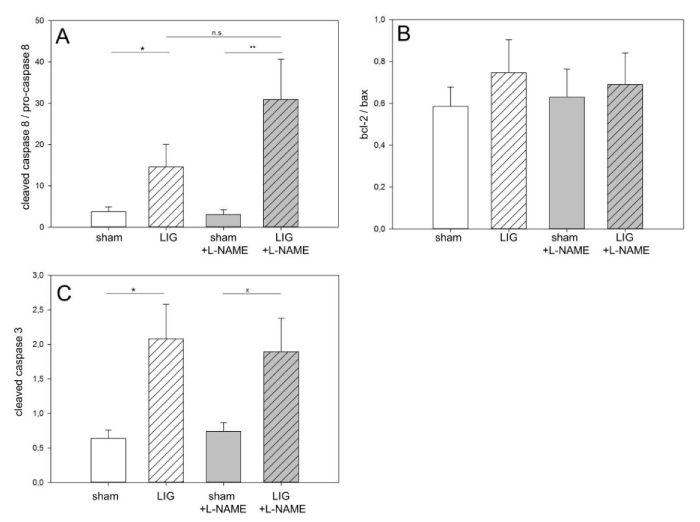
Figure 3
Remote myocardium, apoptosis execution. A: Apoptosis execution assessed by M30 immunohistochemistry was significantly increased in LIG animals. No significant increase was observed in post operation L-NAME treated LIG animals. Compared to native LIG animals, L-NAME treatment caused a significant reduction of the M30-positive cell count.
B: Similarly, apoptosis execution assessed by the TUNEL method was significantly increased in LIG treated animals. Again, no significant increase was observed in post operation L-NAME treated LIG animals.
C + D: Representative visual fields of M30 (C) and TUNEL (D) immunohistochemistry. M30, caspase 3 cleaved ceratin-18 neo-epitope antibody. TUNEL, Terminal Deoxy Nucleotidyl Transferase (TdT)-mediated dUTP nick end-labeling. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending Coronary Artery (LAD). sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Statistical annotations: n.s., not significant;**p = 0.01; ***p = 0.001. n=6-8 per group.
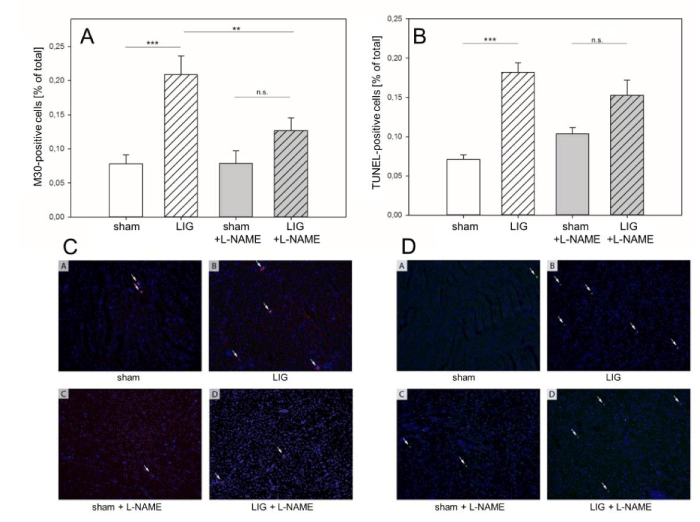
Figure 4
Remote myocardium, fibrotic remodeling. A: LIG treatment caused a significant increase of total fibrotic content in the LV posterior (i.e. remote) myocardium. Post operation L-NAME treatment prevented fibrotic remodeling of the LV posterior myocardium.
B: Representative visual fields of picrosirius red stainings of cryo-sections from the LV posterior myocardium. Treatment groups and labeling as in (A). L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending Coronary Artery (LAD). sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Statistical annotations: *p = 0.05; **p = 0.01. n=6-8 per group.
Discussion
In this study, we show that para-clinical and clinical HF indices were significantly ameliorated by the inhibition of NOS using the specific NOS-inhibitor L-NAME for a treatment period of 14 days after experimental MI in rats in vivo. Moreover, in the remote non-ischemic myocardium L-NAME treatment prevented the induction of apoptosis at the level of effector caspase 3 activation and resulted in significantly less apoptosis execution. Additionally and in consequence, L-NAME treatment prevented fibrotic remodeling of the remote myocardium. On the molecular basis, our data further suggest that the observed beneficial effects of L-NAME treatment are the result of favorable influences on the eNOS ser1177/thre495 phosphorylation ratio with significantly less thr495 phosphorylation in L-NAME treated animals.
Initially, acute MI leads to worsening of cardiac systolic function and typically is followed by a complex sequence of structural changes in the left ventricle, commonly referred to as post-infarction ventricular remodeling [3-5]. These structural changes include cardiac hypertrophy, necrosis and fibrosis and commonly result in progressive chamber dilatation [3-5,7,35]. Recent studies demonstrate that progressive apoptotic loss of cardiomyocytes in the remote myocardium distant from the infarct area crucially contributes to and nourishes this remodeling process [7,25,36]. However, only little is known about the regulation of apoptosis in this zone. The present study sheds light into the regulation of apoptosis in this remote, non-ischemic myocardial zone. Even more, a novel molecular mechanism was identified and the potential reversibility of structural changes and clinically overt HF demonstrated using a therapeutic strategy.
The rat MI animal model used in this study had been well established in our laboratory for years and LAD ligation resulted in reliable and reproducible infarcts of the anterior LV wall [24-27] (For representative images of these study animals, refer to (Figure 1A)). Remarkably, while clinical and para-clinical HF indices improved with L-NAME treatment, these effects were not yet attributable to a measurable recovery of LV systolic function at 14 days. It therefore, may be hypothesized that an improvement of LV diastolic function is most likely the cause for the observed amelioration of HF indices because L-NAME treatment prevented fibrotic remodeling of the remote LV myocardium. Of note, a reduction of myocardial fibrotic remodeling accompanied by significant reductions of NT-proANP serum levels after chronic L-NAME treatment for 2 months was lately shown in a caveolin-1 cardiomyopathy mouse model in vivo, paralleling substantial improvements in diastolic heart function [31].
Significant apoptosis induction was observed via the extrinsic pathway in the remote myocardium of LIG control animals, as expected from previous studies [25]. Notably, L-NAME treatment prevented significantly the induction of apoptosis at the level of the central effector caspase 3. At this level, the signals of the extrinsic and the intrinsic pathways converge [37]. Remarkably, this translated into significantly less apoptosis execution as assessed by M30 immunohistochemistry and TUNEL assay, respectively, and ultimately resulted in less fibrotic remodeling of the remote LV myocardium.
Among other trigger factors, an excess of nitrosative stress is a well-known and potent inductor of myocardial apoptosis [11,12,15]. Differential regulation of endothelial NOS (eNOS) activity is achieved by phosphorylation of the two most important regulatory phosphorylation sites at ser1177 and thr495 [15,19]. Increased phosphorylation at ser1177 is believed to be beneficial as it results in increased NO production with subsequent vasodilatation and anti-inflammatory properties [15,38]. On the other hand, an increase of thr495 phosphorylation is believed to be harmful as it increases eNOS uncoupling potential and promotes improper electron transfer to native oxygen. This increases the formation of superoxide anions which can react with NO to form highly reactive peroxynitrite, resulting an excess of nitrosative stress [15,19]. In this study, we show that L-NAME treatment effectively prevented eNOS phosphorylation at thr495 in LIG animals and that this translated into a preservation of a balanced eNOS ser1177/thr495 phosphorylation ratio in these animals. However, aside from excess phosphorylation at thr495, other factors are known to increase eNOS uncoupling potential, e.g., lack of the essential eNOS cofactor tetrahydrobiopterin (BH4) [14,20,39,40]. We recently demonstrated in vivo that BH4 supplementation resulted in less nitrosative stress and in a reduction of apoptosis in the remote myocardium after MI. Therefore, we conclude that eNOS phosphorylation ratio is not the sole regulatory switch of this tightly controlled key enzyme in the remote ventricular myocardium. Other confounding factors must be considered.
There are some limitations of this study that need to be considered. First, L-NAME is widely used as a specific NOS inhibitor, yet without isoform specificity [41]. However, eNOS is the leading NOS isoform in the heart [18,42]. Therefore and because of the lack of isoform specific NOS inhibitors for in vivo experiments, we believe that it is reasonable to focus on the assessment of eNOS functional status when evaluating L-NAME treatment effects in the myocardium. A second limitation of this study refers to the study design with respect to treatment initiation. To our knowledge, to date no data exist on the timely management of NOS inhibition in the setting of myocardial infarction, i.e. when to initiate therapy for how long and when to stop treatment. For the purpose of this study we sought to resemble clinical reality as close as possible. Therefore, L-NAME treatment was administered as soon as possible after MI immediately when animals awoke from anesthesia. Using this treatment regime, L-NAME treatment for 14 days could not completely prevent the induction and execution of apoptosis in the remote, non-ischemic myocardium. We believe this is most likely due to the contribution of eNOS-independent pro-apoptotic factors. Also, additional studies are warranted to assess whether a prolonged L-NAME treatment period after MI results in more pronounced beneficial effects in terms of apoptosis and fibrosis prevention. Although the investigational focus of this study was on the regulation of apoptosis in the remote myocardium after MI, we acknowledge that, more recently, besides apoptosis the phenomena of autophagy and necroptosis also have been implicated in the ventricular remodeling process of the non-ischemic myocardium, are subject to extensive ongoing research [43-47], yet have not been addressed in our study.
Translational implications of the study
Our data suggest that harmful eNOS activation by differential phosphorylation with increased eNOS-phosphorylation at thr495 mediates apoptosis induction, execution and fibrotic remodeling in the remote non-infarcted LV myocardium. Treatment with the NOS-inhibitor L-NAME restored eNOS ser1177/thr495 phosphorylation ratio and reduced apoptosis induction, execution, and fibrotic remodeling. Moreover, L-NAME treatment ameliorated clinical and para-clinical HF indices, most likely due to an improvement in diastolic heart function. Therefore, the results presented in this in-vivo study strengthen the evidence that influencing eNOS signaling might become an important pharmacological target for the development of novel tailored therapies to prevent the development of HF due to progressive myocardial remodeling after MI.
Figure 5
Remote myocardium, eNOS phosphorylation. A: LIG treatment significantly increased eNOS phosphorylation at threonine495. L-NAME treatment prevented excessive eNOS phosphorylation at threonine495 in LIG animals.
B: eNOS serine1177/threonine495 phosphorylation ratio was significantly reduced in LIG animals. Post operation L-NAME treatment preserved a balanced serine1177/threonine495 eNOS phosphorylation ratio. (p)-eNOS, (phospho)-endothelial nitric oxide synthase. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending Coronary Artery (LAD). sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Statistical annotations: *p = 0.05. n=10-11 per group. Representative western blots may be viewed in the online data supplement (Figure A3).
References
- Murray CJL, Lopez AD. Measuring the Global Burden of Disease. N Engl J Med. 2013; 369: 448-457.
- Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016; 13: 368-378.
- Gajarsa JJ, Kloner RA. Left ventricular remodeling in the post-infarction heart: a review of cellular, molecular mechanisms, and therapeutic modalities. Heart Fail Rev. 2011; 16: 13-21.
- Van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013; 123: 37-45.
- Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990; 81: 1161-1172.
- Tanai E, Frantz S. Pathophysiology of Heart Failure. Compr Physiol. 2015; 6: 187-214.
- Baldi A, Abbate A, Bussani R, Patti G, Melfi R, Angelini A, et al. Apoptosis and post-infarction left ventricular remodeling. J Mol Cell Cardiol. 2002; 34: 165-174.
- Kang PM, Izumo S. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol Med. 2003; 9: 177-182.
- Konstantinidis K, Whelan RS, Kitsis RN. Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol. 2012; 32: 1552-1562.
- Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, et al. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001; 280: H2726-H2731.
- Green DR, Llambi F. Cell Death Signaling. Cold Spring Harb Perspect Biol. 2015; 7.
- Rossi D, Gaidano G. Messengers of cell death: apoptotic signaling in health and disease. Haematologica. 2003; 88: 212-218.
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007; 87: 315-424.
- Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006; 113: 1708-1714.
- Fleming I. Molecular mechanisms underlying the activation of eNOS. Pflugers Arch. 2010; 459: 793-806.
- Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006; 40: 16-23.
- Ebner B, Lange SA, Eckert T, Wischniowski C, Ebner A, Braun-Dullaeus RC, et al. Uncoupled eNOS annihilates neuregulin-1beta-induced cardioprotection: a novel mechanism in pharmacological postconditioning in myocardial infarction. Mol Cell Biochem. 2013; 373: 115-123.
- Kelly RA, Balligand JL, Smith TW. Nitric oxide and cardiac function. Circ Res. 1996; 79: 363-380.
- Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007; 42: 271-279.
- Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009; 297: H1829-H1836.
- Ivanovas B, Zerweck A, Bauer G. Selective and non-selective apoptosis induction in transformed and non-transformed fibroblasts by exogenous reactive oxygen and nitrogen species. Anticancer Res. 2002; 22: 841-856.
- Bauer G. Reactive oxygen and nitrogen species: efficient, selective, and interactive signals during intercellular induction of apoptosis. Anticancer Res. 2000; 20: 4115-4139.
- Saran M, Bors W. Signalling by O2-. and NO.: how far can either radical, or any specific reaction product, transmit a message under in vivo conditions? Chem Biol Interact. 1994; 90: 35-45.
- Schwarz K, Simonis G, Yu X, Wiedemann S, Strasser RH. Apoptosis at a distance: remote activation of caspase-3 occurs early after myocardial infarction. Mol Cell Biochem. 2006; 281: 45-54.
- Simonis G, Wiedemann S, Schwarz K, Christ T, Sedding DG, Yu X, et al. Chelerythrine treatment influences the balance of pro- and anti-apoptotic signaling pathways in the remote myocardium after infarction. Mol Cell Biochem. 2008; 310: 119-128.
- Simonis G, Wiedemann S, Joachim D, Weinbrenner C, Marquetant R, Strasser RH. Stimulation of adenosine A2b receptors blocks apoptosis in the non-infarcted myocardium even when administered after the onset of infarction. Mol Cell Biochem. 2009; 328: 119-126.
- Wiedemann S, Wessela T, Schwarz K, Joachim D, Jercke M, Strasser RH, et al. Inhibition of anti-apoptotic signals by Wortmannin induces apoptosis in the remote myocardium after LAD ligation: evidence for a protein kinase C-delta-dependent pathway. Mol Cell Biochem. 2013; 372: 275-283.
- Wagner C, Tillack D, Simonis G, Strasser RH, Weinbrenner C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol Cell Biochem. 2010; 339: 135-147.
- Guide for the Care and Use of Laboratory Animals. National Academies Press (US) 2011; 8th edition.
- St John SM, Otterstat JE, Plappert T, Parker A, Sekarski D, Keane MG, et al. Quantitation of left ventricular volumes and ejection fraction in post-infarction patients from biplane and single plane two-dimensional echocardiograms. A prospective longitudinal study of 371 patients. Eur Heart J. 1998; 19: 808-816.
- Wunderlich C, Schober K, Kasper M, Heerwagen C, Marquetant R, Ebner B, et al. Nitric oxide synthases are crucially involved in the development of the severe cardiomyopathy of caveolin-1 knockout mice. Biochem Biophys Res Commun. 2008; 377: 769-774.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680-685.
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979; 76: 4350-4354.
- Vogel B, Siebert H, Hofmann U, Frantz S. Determination of collagen content within picrosirius red stained paraffin-embedded tissue sections using fluorescence microscopy. MethodsX. 2015; 2: 124-134.
- Opie LH, Commerford PJ, Gersh BJ, Pfeffer MA. Controversies in ventricular remodelling. Lancet. 2006; 367: 356-367.
- Cheng W, Kajstura J, Nitahara JA, Li B, Reiss K, Liu Y, et al. Programmed myocyte cell death affects the viable myocardium after infarction in rats. Exp Cell Res. 1996; 226: 316-327.
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35: 495-516.
- Ebner B, Ebner A, Reetz A, Bohme S, Schauer A, Strasser RH, et al. Pharmacological postconditioning by bolus injection of phosphodiesterase-5 inhibitors vardenafil and sildenafil. Mol Cell Biochem. 2013; 379: 43-49.
- Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004; 24: 413-420.
- Wever RM, van DT, van Rijn HJ, de GF, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. 1997; 237: 340-344.
- Griffith OW, Kilbourn RG. Nitric oxide synthase inhibitors: amino acids. Methods Enzymol. 1996; 268: 375-392.
- Ziolo MT, Kohr MJ, Wang H. Nitric oxide signaling and the regulation of myocardial function. J Mol Cell Cardiol. 2008; 45: 625-632.
- Gupta SK, Foinquinos A, Thum S, Remke J, Zimmer K, Bauters C, et al. Preclinical Development of a MicroRNA-Based Therapy for Elderly Patients With Myocardial Infarction. J Am Coll Cardiol. 2016; 68: 1557-1571.
- Liu J, Wu P, Wang Y, Du Y, AN, Liu S, et al. Ad-HGF improves the cardiac remodeling of rat following myocardial infarction by upregulating autophagy and necroptosis and inhibiting apoptosis. Am J Transl Res. 2016; 8: 4605-4627.
- Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015; 22: 367-376.
- Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014; 103: 206-216.
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010; 11: 700-714.
Figure 1A
Evaluation of infarct size, area at risk and viable myocardium at 14 days after induction of anterior Myocardial Infarction (MI. Representative images are shown. MI was induced by ligation of the left anterior descending coronary artery (LAD). As expected, no infarct was obvious in sham treated control animals. LIG treated animals, however, showed marked infarct demarcation. Left column, native heart slices. Right column, corresponding UV-light microscopy images; infarct zone (pink) - resembling propidium iodide DNA intercalation, viable myocardium (green) - resembling fluorescent microspheres accumulation, area at risk - infarct zone + non-fluorescent areas localized adjacent to and within the infarct zone with the latter resembling LAD supplied but not necrotic myocardium. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing LAD ligation. sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. n = 4 per group.
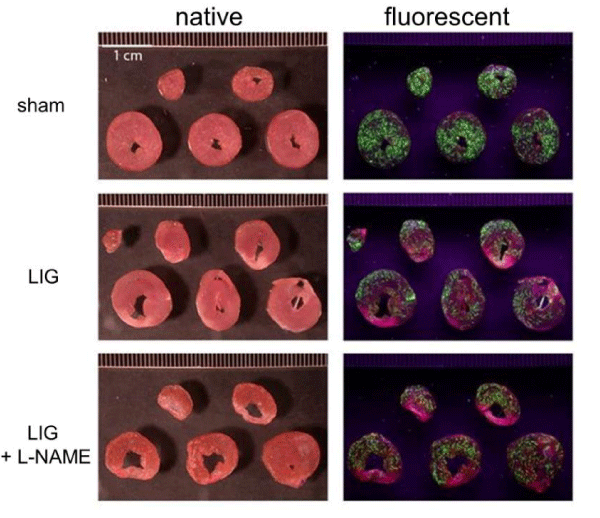
Figure 2A
Representative western blots corresponding to bar graphs in Figure 2. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending coronary artery (LAD). Sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Molecular weights given in kilo Daltons (kDa). n = 10-11 per group.
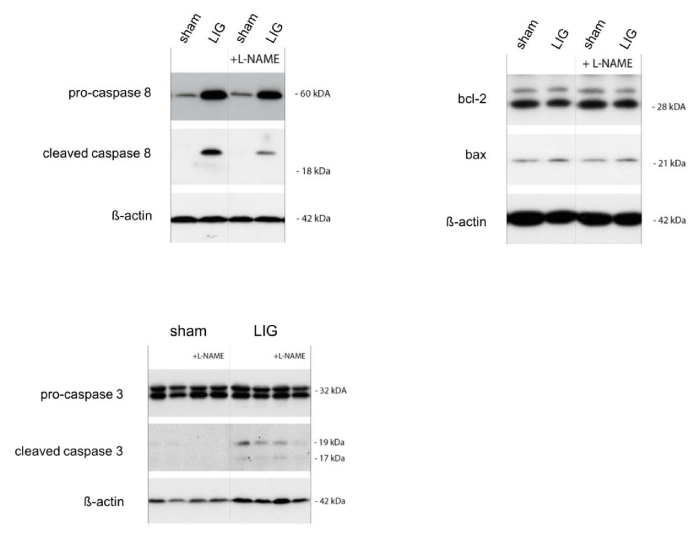
Figure 3A
Representative western blots corresponding to bar graphs in Figure 5. (p)-eNOS, (phospho)-endothelial nitric oxide synthase. L-NAME, N?-Nitro-L-arginine methyl ester. LIG, treatment group experiencing chronic ligation of the Left Anterior Descending Coronary Artery (LAD). Sham, treatment group operated and treated identically compared to LIG animals except for ligation of the LAD. Molecular weights given in kilo Daltons (kDa). n = 10-11 per group.
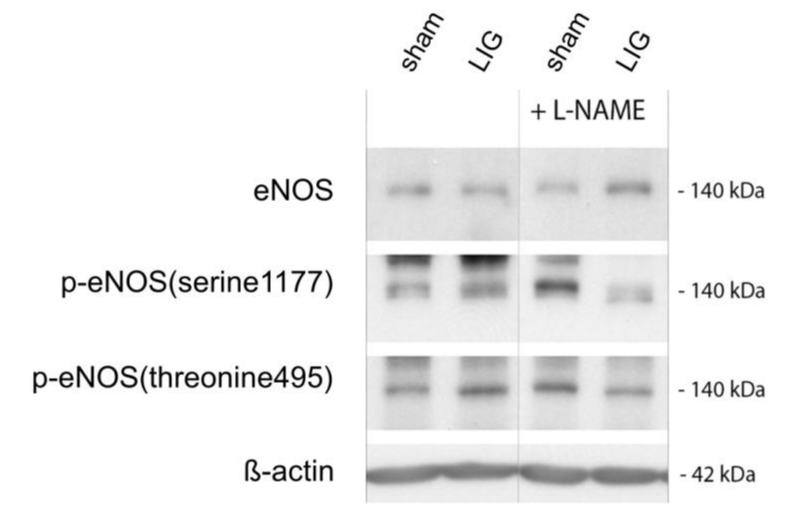
Table 1A
| Table 1A: Synopsis of the primary and secondary antibodies used | |||
| (p)-eNOS, (phospho)-Endothelial Nitric Oxide Synthase. mAB: Monoclonal Antibody. | |||
| Antibody | Host | Company | Dilution |
| Primary antibodies | |||
| anti-caspase 8 (D35G2) | rabbit | CellSignaling, Danvers, USA | 1:500 |
| anti-cleavedcaspase 8 (Asp387) | rabbit | CellSignaling, Danvers, USA | 1:500 |
| anti-cleavedcaspase 3 (Asp175) | rabbit | CellSignaling, Danvers, USA | 1:250 |
| anti-bax (PC66) | rabbit | Calbiochem, Schwalbach, GER | 1:500 |
| anti-bcl-2 (N-19) | rabbit | CellSignaling, Danvers, USA | 1:500 |
| anti-eNOS | rabbit | CellSignaling, Danvers, USA | 1:500 |
| anti-phospho-eNOS(serine1177) | rabbit | CellSignaling, Danvers, USA | 1:250 |
| anti-phospho-eNOS(threonine495) | rabbit | CellSignaling, Danvers, USA | 1:250 |
| anti- -actin | mouse | Santa Cruz, Heidelberg, GER | 1:1.000 |
| Secondary antibodies | |||
| IgG HRP-linked anti-rabbit mAB | goat | CellSignaling, Danvers, USA | 1:2.000 |
| IgG HRP-linked anti-mouse mAB | sheep | CellSignaling, Danvers, USA | 1:2.000 |
Authors submit all Proposals and manuscripts via Electronic Form!


























