

Keiko Kawauchi* Wataru Sugimoto1, Katsuhiko Itoh1, Toshiya Kotari, Alvin K. Guo, Takahiro Ebata and Hiroaki Hirata
Wataru Sugimoto1#, Katsuhiko Itoh1#, Toshiya Kotari1#, Alvin K. Guo2, Takahiro Ebata1, Hiroaki Hirata3, Keiko Kawauchi1,4*
1Frontiers of Innovative Research in Science and Technology, Konan University, Japan
2Cancer & Stem Cell Biology Program, Duke-NUS Medical School, Singapore
3Nagoya University Graduate School of Medicine, Japan
4Department of Molecular Oncology, Institute for Advanced Medical Sciences, Japan
#These authors contributed equally to this work
*Address for Correspondence:Keiko Kawauchi, Frontiers of Innovative Research in Science and Technology (FIRST), Konan university, 7-1-20 Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan, Tel: +81-78-303-1346; Fax: +81-78-303-1549; E-mail: kawauchi@center.konan-u.ac.jp
Dates: Submitted: 19 May 2017; Approved: 23 May 2017; Published: 24 May 2017
Citation this article: Sugimoto W, Itoh K, Kotari T, Guo AK, Ebata T, et al. NF-κB Prevents Oncogenic Ras-Induced β-Actin Cleavage in p53-Deficient Cells. Int J Cancer Cell Biol Res. 2017; 2(1): 014-018.
Copyright: © 2017 Sugimoto W, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: p53; NF-κB; Actin; Mitochondria; HtrA2/ Omi; Lamellipodia
Abstract
Tumor cell characteristics, including invasiveness, are influenced by the tumor suppressor p53. We recently reported that p53 diminishes oncogenic Ras-driven cell invasion by promoting β-actin cleavage, which is mediated by the serine protease high-temperature requirement A2 (HtrA2/ Omi). In this study, we showed that nuclear factor (NF)-κB is responsible for eliminating Ras - induced β-actin cleavage in p53 - deficient cells. In p53 knockout (p53-/-) Mouse Embryonic Fibroblasts (MEFs), depletion of the NF-κB component p65/ RelA using its specific shRNA caused β-actin cleavage in response to expression of oncogenic Ras. Knockdown of p65/ RelA enhanced the mitochondrial translocation of p38 MAPK, a critical step in HtrA2/Omi activation. Expression of mitochondria-targeting signal-tagged p65/ RelA diminished β-actin cleavage in p65/ RelA-knockdown cells. Our results provide novel insights into the mechanism underlying contribution of NF-κB to malignancy of Ras-mutated cells with dysfunctional p53.
Introduction
Ras controls fundamental cell behaviors, such as proliferation and survival, by regulating downstream signaling that includes the Raf-MEK-MAPK and PI3K-Akt pathways [1-3]. On the contrary, aberrant activation of Ras accompanied with its gene mutations is observed in several types of cancers. When oncogenic mutations occur in the RAS gene, the tumor suppressor p53 plays an essential role in determining cell behaviors. Expression of oncogenic Rasin normal cells triggers DNA damage responses and induces apoptosis or senescence by induction of p53-dependent gene expression [4,5]. In contrast, oncogenic Ras induces cellular transformation when the ability of p53 to prevent tumorigenesis is ablated as a consequence of its gene mutation or inactivation [6]. Furthermore, while oncogenic Ras promotes invasion and metastasis in transformed cells, these effects of Ras are also diminished by p53 [7]. Thus, Ras–induced tumorigenesis and tumor progression are both dependent on the status of p53.
Recently, we have revealed a mechanism underlying suppression of Ras-driven cell invasion by p53. Oncogenic Ras stimulates cleavage of β-Actin by the mitochondrial protease, high-temperature requirement A2 (HtrA2; also known as Omi), which depends on the cytoplasmic p53 [8]. The Ras-induced cleavage of β-Actin eliminates the formation of lamellipodia, actin-based membrane protrusions crucial for cancer cell invasion [9,10].
It has been well documented that the transcription factor NF-κB plays an essential role in Ras-induced cellular transformation by p53 dysfunction [6,11,12]. Oncogenic Ras promotes the metabolic shift from Oxidative Phosphorylation (OXPHO) to aerobic glycolysis, known as the Warburg effect [13], which is, at least in part, attributed to NF-κB-induced expression of Glucose Transporter (GLUT) 3 [6]. Furthermore, NF-κB-mediated gene expression is required for Epithelial-Mesenchymal Transition (EMT), which is involved in the early steps of metastasis, in Ras-transformed cells [14]. p53 suppresses the NF-κB-mediated gene expression by attenuating both IκB kinase (IKK) activity and interaction of NF-κB with its co-activator p300/CBP [6,15-17]. While NF-κB translocates to the mitochondria to repress the expression of OXPHO-related mitochondrial genes, p53 prevents the translocation of NF-κB to the mitochondria by inhibiting the interaction of NF-κB with the mitochondrial heat shock protein (mt-HSP70; also known as Mortalin) [18]. However, the functional role of mitochondrial NF-κB in oncogenic Ras-expressing cells remains unclear.
Here, we show that in the absence of p53, knock down of the NF-κB component, p65/ RelA, increases β-Actin cleavage and decreases lamellipodia formation in cells expressing oncogenic Ras. Ectopic expression of a mitochondria-targeting form of p65/ RelA, in turn, reduces cleavage of β-Actin. Our results suggest that NF-κB may promote oncogenic Ras-induced invasion of p53-deficient cells by facilitating the formation of lamellipodia through reduction of β-Actin cleavage.
Materials and Methods
Cell culture and retroviral infection
p53–/– MEFs were prepared as previously described [6] and infected following 3 passages. Cells were cultured in Dulbecco’s modified Eagle’s medium (Nissui) supplemented with 10% FBS. We performed all experiments within two weeks after selection to avoid genetic abnormalities acquired during prolonged culture. Retroviral infection was performed as previously described [19]. Infected cells were selected using hygromycin (300 µg/ ml) and puromycin (1.5 µg/ ml) for 3 days.
Plasmids
pSuper retro p65 puro was cloned p65 target sequence [6], 5′-GAAGAAGAGTCCTTTCAAT-3′ into a pSuper retro puro (Oligoengine, Seattle, WA). pBabe Ha-RasV12 with a hygromycin selection marker were used [6]. The Mito-DsRed expression vector was obtained from Clontech Laboratories Inc. To generate a pcDNA3-Mito-Flag-p65 vector, the mitochondrial targeting sequence from subunit VIII of human cytochrome c oxidase was fused to the 5′-end of the Flag-tagged p65.
Antibodies and materials
Anti-p38 rabbit polyclonal (C-20, Santa Cruz Biotechnology), p38 rabbit polyclonal (Cell Signaling Technology), anti-HtrA2/ Omi goat polyclonal (V-17, Santa Cruz Biotechnology), anti-β-Actin mouse monoclonal (ACTBD11B7, Santa Cruz Biotechnology), anti-VDAC rabbit polyclonal (Cell Signaling), and anti-α-tubulin mouse monoclonal (DM1A, Sigma) were used for immunoblot analysis. Anti-Flag mouse monoclonal (M2, Sigma) antibodies were used for immunoblot and immunofluorescence analyses. Rabbit (H-191, Santa Cruz Biotechnology) was used for immunofluorescence analysis.
Immunoblot analysis
To obtain total cell lysates, cells were solubilized with ice-cold lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% SDS, 10 mM EDTA, 1 mM Na3VO4, 10 mM NaF, protease inhibitor cocktail [Nakarai Tesque]). Lysates were sonicated and centrifuged at 20,000 x g for 15 min. The supernatants were used as total cell lysates. To obtain mitochondria and cytosol fractions, a mitochondria isolation kit (QIAGEN) was used according to the manufacturer’s protocol. The isolated mitochondrial fraction was solubilized with ice-cold lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% Triton X-100, 10 mM EDTA, 1 mM Na3VO4, 10 mM NaF, protease inhibitor cocktail) and then centrifuged at 20,000 x g for 15 min. The supernatants were used as mitochondrial proteins. For immunoblotting against β-Actin, cells were solubilized with an SDS sample buffer. The lysates were subjected to Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE).
Immunofluorescence
Cells were fixed with 4% PFA, permeabilized with 0.2% Triton X-100, and then blocked with 2% BSA in PBS. Alexa Fluor 488-conjugated goat anti-rabbit IgG (Molecular Probes) and Alexa Fluor 546-conjugated goat anti-mouse IgG were used as secondary antibodies. Alexa Fluor 594-conjugated phalloidin (Molecular Probes) were used to stain F-actin. Images were acquired using a confocal microscope (LSM700; Zeiss) and then analyzed with ImageJ software (NIH).
Monitoring mitochondrial membrane potential
Monitoring mitochondrial membrane potential was performed as previously described [8]. Acquired images using a confocal microscope (LSM700; Zeiss) were cropped and fluorescence intensities were quantified using Image J software (version 1.45f).
Statistical analysis
Data were analyzed by unpaired Student’s t-test.
Results and Discussion
Lamellipodia formation has been shown to be suppressed upon expression of oncogenic Ras in cells bearing wild-type p53, but not in p53-deficient cells [8]. We found that p65/ RelA knock down diminished lamellipodia formation in p53–/– MEFs expressing oncogenic Ras (Ha-RasV12) (Figure 1A). Since oncogenic Ras-induced β-Actin cleavage by HtrA2/ Omi impairs lamellipodia formation, we further examined whether p65/ RelA knock down stimulates β-Actin cleavage in p53–/– MEFs. Immunoblotting showed the fragment of β-Actin in cells expressing both Ha-RasV12 and p65/ RelA-shRNA, while Ha-RasV12 expression or p65/ RelA knock down alone was not sufficient for induction of β-Actin cleavage (Figure 1B). These results suggest that NF-κB contributes to lamellipodia formation in Ras-transformed cells by reducing the actin proteolysis.
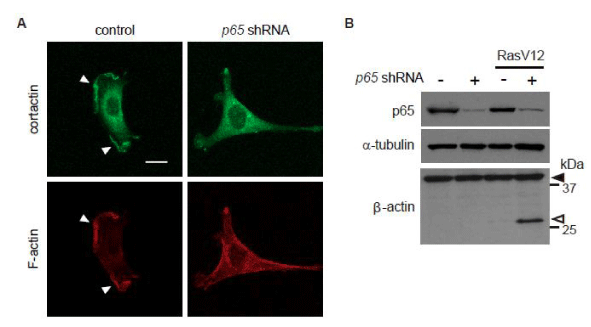 Figure 1: Oncogenic Ras induces β-actin cleavage in p53–/– MEFs expressing 65/RelA shRNA.
Cells were infected with a control or Ha-Ras V12-expressing retrovirus together with or without a p65/RelA shRNA-expressing retrovirus. (A) Confocal images of the cells stained for cortactin (green) as an indicator of lamellipodia or F-actin (red) are shown. Scale bars, 20 µm. Z-stack images with an interval of 1.0µm were obtained using a confocal microscope, and projected images are shown. The white arrowheads point to the edge of the lamellipodia. (B) The levels of p65/RelA and the cleavage of β-actin were evaluated by immunoblot analysis. Black arrowheads indicate full-length β-actin, and white arrowheads indicate cleaved fragment.
Figure 1: Oncogenic Ras induces β-actin cleavage in p53–/– MEFs expressing 65/RelA shRNA.
Cells were infected with a control or Ha-Ras V12-expressing retrovirus together with or without a p65/RelA shRNA-expressing retrovirus. (A) Confocal images of the cells stained for cortactin (green) as an indicator of lamellipodia or F-actin (red) are shown. Scale bars, 20 µm. Z-stack images with an interval of 1.0µm were obtained using a confocal microscope, and projected images are shown. The white arrowheads point to the edge of the lamellipodia. (B) The levels of p65/RelA and the cleavage of β-actin were evaluated by immunoblot analysis. Black arrowheads indicate full-length β-actin, and white arrowheads indicate cleaved fragment.
A decrease in the inner mitochondrial membrane potential (ΔΨm) is involved in the β-Actin cleavage [8]. We, therefore, examined the effect of p65/ RelA knockdown on ΔΨm in p53–/– MEFs expressing Ha-RasV12, and found that ΔΨm was significantly decreased in a subset of mitochondria (Figure 2A and B). A decrease in ΔΨm causes mitochondrial translocation of p38 MAPK, which leads to HtrA2/ Omi activation to cleave β-Actin [8]. Consistently, the amount of p38 MAPK in the mitochondrial fraction was higher in p65/ RelA knock down cells than in control cells, even though the amount of HtrA2/ Omi in cytosol was not significantly different between these cells (Figure 2C). Taken together, NF-κB is likely to suppress HtrA2/ Omi-mediated β-Actin cleavage by maintaining ΔΨm to eliminate mitochondrial translocation of p38 MAPK.
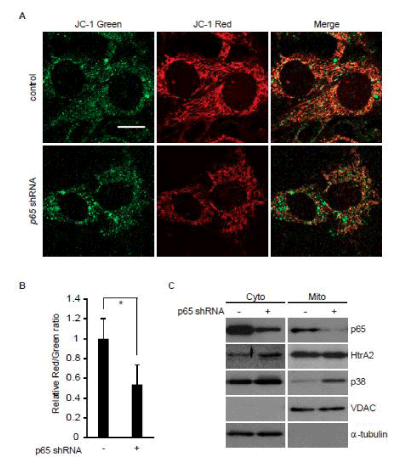 Figure 2: p65/RelA knockdown enhances mitochondrial translocation of p38 MAPK in p53–/– MEFs expressing Ha-Ras V12. Cells were infected with Ha-Ras V12-expressing retrovirus together with or without a p65/RelA shRNA-expressing retrovirus. (A) Confocal images of cells stained with JC-1. Red fluorescence (J-aggregate) and green fluorescence (monomer) are shown. Scale bars, 20 µm. (B) The ratio of red fluorescence to green fluorescence, which is correlated with mitochondrial membrane potential, at peripheral mitochondria was quantified from images in A. (C) Following subcellular fractionation of the cytosol (Cyto) and mitochondria (Mito), the distribution of p65/RelA, p38 MAPK, and HtrA2/Omi was evaluated by immunoblot analysis. VDAC and -tubulin were used as mitochondrial and cytosolic markers, respectively.
Figure 2: p65/RelA knockdown enhances mitochondrial translocation of p38 MAPK in p53–/– MEFs expressing Ha-Ras V12. Cells were infected with Ha-Ras V12-expressing retrovirus together with or without a p65/RelA shRNA-expressing retrovirus. (A) Confocal images of cells stained with JC-1. Red fluorescence (J-aggregate) and green fluorescence (monomer) are shown. Scale bars, 20 µm. (B) The ratio of red fluorescence to green fluorescence, which is correlated with mitochondrial membrane potential, at peripheral mitochondria was quantified from images in A. (C) Following subcellular fractionation of the cytosol (Cyto) and mitochondria (Mito), the distribution of p65/RelA, p38 MAPK, and HtrA2/Omi was evaluated by immunoblot analysis. VDAC and -tubulin were used as mitochondrial and cytosolic markers, respectively.
While p53 reportedly suppresses mitochondrial translocation of NF-κB [18], NF-κB was indeed found in the mitochondrial fraction in p53–/– MEFs expressing Ha-RasV12 (Figure 2C). We, therefore, hypothesized that mitochondrial NF-κB might prevent p38 MAPK translocation into mitochondria and concomitant β-Actin cleavage in oncogenic Ras-expressing p53-deficient cells. To test this hypothesis, the mitochondrial targeting peptide-fused flag-tagged p65 (Mito-Flag-p65) was expressed in p65/ RelA-knockdown p53–/– MEFs expressing Ha-RasV12. While Mito-Flag-p65 was localized at the mitochondria (Figure 3A), the amount of p38 MAPK in the mitochondrial fraction was unexpectedly not affected by Mito-Flag-p65 expression (Figure 3B). However, Mito-Flag-p65 expression decreased β-Actin cleavage (Figure 3C). These results suggest that elimination of oncogenic Ras-induced β-Actin cleavage by mitochondrial NF-κB is not mediated by the regulation of p38 MAPK translocation into mitochondria. Thus, in addition to its well-established role as a transcriptional factor, NF-κB regulates proteolysis of the cytoskeletal element under the Ras-transformed condition.
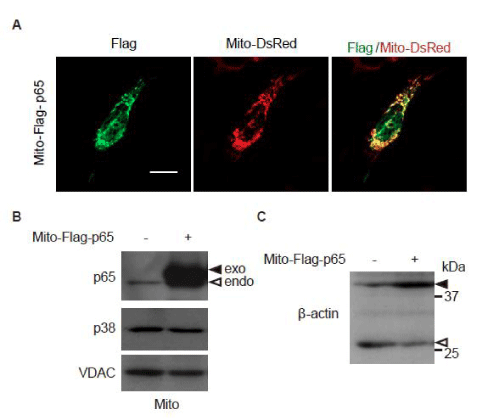 Figure 3: Expression of p65/RelA, localized in the mitochondria, attenuates -actin cleavage in p65/RelA-knockdown p53–/– MEFs expressing Ha-Ras V12. Cells were infected with Ha-Ras V12-expressing retrovirus together with a p65/RelA shRNA-expressing retrovirus. (A) The cells were transfected with Mito-Flag-p65 expression vector together with Mito-DsRed expression vector to visualize mitochondria. Confocal images of cells stained with anti-Flag antibody for Mito-Flag-p65 (green) and Mito-DsRed (red) are shown. Z-stack images with an interval of 1.0 µm were obtained using a confocal microscope, and projected images are shown. Scale bars, 20 µm. (B) and (C) The cells were transfected with control or Mito-Flag-p65 expression vector. (B) Following subcellular fractionation of the mitochondria (Mito), the distribution of p38 MAPK was evaluated by immunoblot analysis. VDAC was used as a mitochondrial marker. Black arrows indicate exogenous p65/RelA, and white arrow indicates endogenous p65/RelA. (C) The cleavage of β-actin was evaluated by immunoblot analysis. Black arrowheads indicate full-length β-actin, and white arrowheads indicate the cleaved fragment.
Figure 3: Expression of p65/RelA, localized in the mitochondria, attenuates -actin cleavage in p65/RelA-knockdown p53–/– MEFs expressing Ha-Ras V12. Cells were infected with Ha-Ras V12-expressing retrovirus together with a p65/RelA shRNA-expressing retrovirus. (A) The cells were transfected with Mito-Flag-p65 expression vector together with Mito-DsRed expression vector to visualize mitochondria. Confocal images of cells stained with anti-Flag antibody for Mito-Flag-p65 (green) and Mito-DsRed (red) are shown. Z-stack images with an interval of 1.0 µm were obtained using a confocal microscope, and projected images are shown. Scale bars, 20 µm. (B) and (C) The cells were transfected with control or Mito-Flag-p65 expression vector. (B) Following subcellular fractionation of the mitochondria (Mito), the distribution of p38 MAPK was evaluated by immunoblot analysis. VDAC was used as a mitochondrial marker. Black arrows indicate exogenous p65/RelA, and white arrow indicates endogenous p65/RelA. (C) The cleavage of β-actin was evaluated by immunoblot analysis. Black arrowheads indicate full-length β-actin, and white arrowheads indicate the cleaved fragment.
NF-κB upregulates the expression of integrins αv and β3 [20-22], which are also involved in lamellipodia formation in p53-depleted cells [19]. This together with the results of this study on NF-κB-dependent repression of β-Actin cleavage, indicates that NF-κB is likely to play a prominent role in increasing oncogenic Ras-driven tumor cell malignancy by promoting lamellipodia formation.
Acknowledgements
We thank Dr. Shota Yamauchi for discussion. This work was supported by JSPS KAKENHI Grant Number 26890024and Grant for Collaborative Research from Nagoya University (2614Dj-02b).
References
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011; 11: 761-774. https://goo.gl/4oVCp4
- Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer. 2011; 2: 261-274. https://goo.gl/n7b1dt
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010; 1802: 396-405. https://goo.gl/mwy6lN
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 88: 593-602. https://goo.gl/zYRzEI
- Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier: damage threshold and the concept of 'conditional haploinsufficiency'. Cell Cycle. 2007; 6: 2344-2347. https://goo.gl/A4KAju
- Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008; 10: 611-618. https://goo.gl/IffcpG
- Campbell PM, Der CJ. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin Cancer Biol. 2004; 14: 105-114. https://goo.gl/A9krxL
- Yamauchi S, Hou YY, Guo AK, Hirata H, Nakajima W, Yip AK, et al. p53-mediated activation of the mitochondrial protease HtrA2/Omi prevents cell invasion. J Cell Biol. 2014; 204: 1191-1207. https://goo.gl/TE11uc
- Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis. 2009; 26: 273-287. https://goo.gl/AKhsA8
- Petrie RJ, Yamada KM. At the leading edge of three-dimensional cell migration. J Cell Sci. 2012; 125: 5917-5926. https://goo.gl/oH8IKd
- Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS Jr. Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997; 272: 24113-24116. https://goo.gl/Lvvsni
- Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000; 1470: M55-M62. https://goo.gl/VrY2HF
- Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009; 23: 537-548. https://goo.gl/7Pdl4O
- Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004; 114: 569-581. https://goo.gl/xiVdFi
- Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces NF-kappaB DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun. 2008; 372: 137-141. https://goo.gl/2PFqag
- Kawauchi K, Araki K, Tobiume K, Tanaka N. Loss of p53 enhances catalytic activity of IKKbeta through O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci U S A. 2009; 106: 3431-3436. https://goo.gl/5JgKLD
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999; 19: 3485-3495. https://goo.gl/Tzi2fC
- Johnson RF, Witzel, II, Perkins ND. p53-dependent regulation of mitochondrial energy production by the RelA subunit of NF-kappaB. Cancer Res. 2011; 71: 5588-5597. https://goo.gl/h1le3F
- Guo AK, Hou YY, Hirata H, Yamauchi S, Yip AK, Chiam KH, et al. Loss of p53 enhances NF-kappaB-dependent lamellipodia formation. J Cell Physiol. 2014; 229: 696-704. https://goo.gl/0FJPRd
- Yeh YY, Chiao CC, Kuo WY, Hsiao YC, Chen YJ, Wei YY, et al. TGF-beta1 increases motility and alphavbeta3 integrin up-regulation via PI3K, Akt and NF-kappaB-dependent pathway in human chondrosarcoma cells. Biochem Pharmacol. 2008; 75: 1292-1301. https://goo.gl/qAqKRo
- Jinushi M, Chiba S, Baghdadi M, Kinoshita I, Dosaka-Akita H, Ito K, et al. ATM-mediated DNA damage signals mediate immune escape through integrin-alphavbeta3-dependent mechanisms. Cancer Res. 2012; 72: 56-65. https://goo.gl/KrfUbK
- Hou CH, Yang RS, Hou SM, Tang CH. TNF-alpha increases alphavbeta3 integrin expression and migration in human chondrosarcoma cells. J Cell Physiol. 2011; 226: 792-799. https://goo.gl/LpXFjV
Authors submit all Proposals and manuscripts via Electronic Form!


























